9 Methylation analysis
9.1 Initialization
if (!require("tet.gastru")) {
if (!require("devtools")) {
install.packages("devtools")
}
devtools::install(here("scripts/tet.gastru"))
}## Loading required package: tet.gastru## here() starts at /net/mraid14/export/tgdata/users/aviezerl/temp/test_tet_gastru/tet-gastrulation9.2 Define genomic contexts
9.2.0.4 K4me3/K27me3 peaks
Due to the large size of the ENCODE tracks which generated the CHIP peaks, we supply here also the result of our computation. You can see the details at: scripts/tet.gastru/R/genomic-context.R
define_k4me3_peaks()define_k27me_peaks()define_atac_peaks()
In a nutshell - we aggregate functional annotation using data from ENCODE (Bing Ren’s lab), and from the following paper:
Xiang, Y., Zhang, Y., Xu, Q. et al. Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat Genet 52, 95–105 (2020). https://doi.org/10.1038/s41588-019-0545-1
9.3 Extract methylation data
We will start by extracting all methylation data:
9.3.1 All CpGs
We extract methylation in smoothed over 200bp from each side of the CpG, and not smoothed:
meth_df_comp <- gextract_meth(
c(
comb_tracks,
"e8_5",
"Zhang_Nature_Genetics_2017.Ect_mCG",
"Zhang_Nature_Genetics_2017.End_mCG",
"Zhang_Nature_Genetics_2017.Mes_mCG",
"Zhang_Nature_Genetics_2017.E65Epi_mCG"
),
names = c(comb_names, "e8.5", "ecto", "endo", "meso", "epi"),
extract_meth_calls = TRUE,
d_expand = 200,
annot_tracks = "ctcf"
) %>%
mutate(e7.5.meth = ecto.meth + endo.meth + meso.meth, e7.5.cov = ecto.cov + endo.cov + meso.cov, e7.5 = e7.5.meth / e7.5.cov) %cache_df%
here("output/cpg_meth_expand_200_comp.tsv") %>%
as_tibble()
nrow(meth_df_comp)## [1] 21342750## # A tibble: 6 x 29
## chrom start end ctrl tet_tko e8.5 ecto endo
## 1 chr1 3000573 3000574 1.0000000 0.8333333 0.6666667 0.7333333 0.4583333
## 2 chr1 3000725 3000726 1.0000000 0.8333333 0.5714286 0.6111111 0.4516129
## 3 chr1 3000900 3000901 1.0000000 1.0000000 0.3333333 0.6666667 0.4230769
## 4 chr1 3001345 3001346 0.8888889 0.8333333 0.9600000 0.8888889 0.4000000
## 5 chr1 3001393 3001394 0.8888889 0.8333333 0.9600000 0.8888889 0.4000000
## 6 chr1 3001630 3001631 1.0000000 0.7500000 1.0000000 0.7857143 0.8333333
## meso epi ctrl.cov tet_tko.cov e8.5.cov ecto.cov endo.cov meso.cov
## 1 0.7600000 0.6842105 8 6 6 15 24 25
## 2 0.7307692 0.6190476 8 6 7 18 31 26
## 3 0.6250000 0.5000000 1 2 3 9 26 8
## 4 0.7058824 1.0000000 9 6 25 9 15 17
## 5 0.7058824 1.0000000 9 6 25 9 15 17
## 6 1.0000000 1.0000000 5 4 2 14 12 19
## epi.cov ctrl.meth tet_tko.meth e8.5.meth ecto.meth endo.meth meso.meth
## 1 19 8 5 4 11 11 19
## 2 21 8 5 4 11 14 19
## 3 14 1 2 1 6 11 5
## 4 5 8 5 24 8 6 12
## 5 5 8 5 24 8 6 12
## 6 8 5 3 2 11 10 19
## epi.meth ctcf intervalID e7.5.meth e7.5.cov e7.5
## 1 13 0.6776 1 41 64 0.6406250
## 2 13 0.1325 1 44 75 0.5866667
## 3 7 0.8903 1 22 43 0.5116279
## 4 5 0.7201 1 26 41 0.6341463
## 5 5 0.2538 1 26 41 0.6341463
## 6 8 0.8507 1 40 45 0.8888889meth_df_comp_non_smoothed <- gextract_meth(
c(
comb_tracks,
"e8_5",
"Zhang_Nature_Genetics_2017.Ect_mCG",
"Zhang_Nature_Genetics_2017.End_mCG",
"Zhang_Nature_Genetics_2017.Mes_mCG",
"Zhang_Nature_Genetics_2017.E65Epi_mCG"
),
names = c(comb_names, "e8.5", "ecto", "endo", "meso", "epi"),
extract_meth_calls = TRUE,
d_expand = 200,
annot_tracks = "ctcf"
) %>%
mutate(e7.5.meth = ecto.meth + endo.meth + meso.meth, e7.5.cov = ecto.cov + endo.cov + meso.cov, e7.5 = e7.5.meth / e7.5.cov) %cache_df%
here("output/cpg_meth_comp.tsv") %>%
as_tibble()
nrow(meth_df_comp_non_smoothed)## [1] 21342750## # A tibble: 6 x 29
## chrom start end ctrl tet_tko e8.5 ecto endo
## 1 chr1 3000573 3000574 1.0000000 0.8333333 0.6666667 0.7333333 0.4583333
## 2 chr1 3000725 3000726 1.0000000 0.8333333 0.5714286 0.6111111 0.4516129
## 3 chr1 3000900 3000901 1.0000000 1.0000000 0.3333333 0.6666667 0.4230769
## 4 chr1 3001345 3001346 0.8888889 0.8333333 0.9600000 0.8888889 0.4000000
## 5 chr1 3001393 3001394 0.8888889 0.8333333 0.9600000 0.8888889 0.4000000
## 6 chr1 3001630 3001631 1.0000000 0.7500000 1.0000000 0.7857143 0.8333333
## meso epi ctrl.cov tet_tko.cov e8.5.cov ecto.cov endo.cov meso.cov
## 1 0.7600000 0.6842105 8 6 6 15 24 25
## 2 0.7307692 0.6190476 8 6 7 18 31 26
## 3 0.6250000 0.5000000 1 2 3 9 26 8
## 4 0.7058824 1.0000000 9 6 25 9 15 17
## 5 0.7058824 1.0000000 9 6 25 9 15 17
## 6 1.0000000 1.0000000 5 4 2 14 12 19
## epi.cov ctrl.meth tet_tko.meth e8.5.meth ecto.meth endo.meth meso.meth
## 1 19 8 5 4 11 11 19
## 2 21 8 5 4 11 14 19
## 3 14 1 2 1 6 11 5
## 4 5 8 5 24 8 6 12
## 5 5 8 5 24 8 6 12
## 6 8 5 3 2 11 10 19
## epi.meth ctcf intervalID e7.5.meth e7.5.cov e7.5
## 1 13 0.6776 1 41 64 0.6406250
## 2 13 0.1325 1 44 75 0.5866667
## 3 7 0.8903 1 22 43 0.5116279
## 4 5 0.7201 1 26 41 0.6341463
## 5 5 0.2538 1 26 41 0.6341463
## 6 8 0.8507 1 40 45 0.88888899.4 Distribution of TET-TKO vs WT methylation (Figure 5B)
options(repr.plot.width = 7, repr.plot.height = 7)
min_cov <- 20
plot_smooth_cpg_scatter(meth_df_comp, "ctrl", "tet_tko", min_cov, "Control", "Tet-TKO")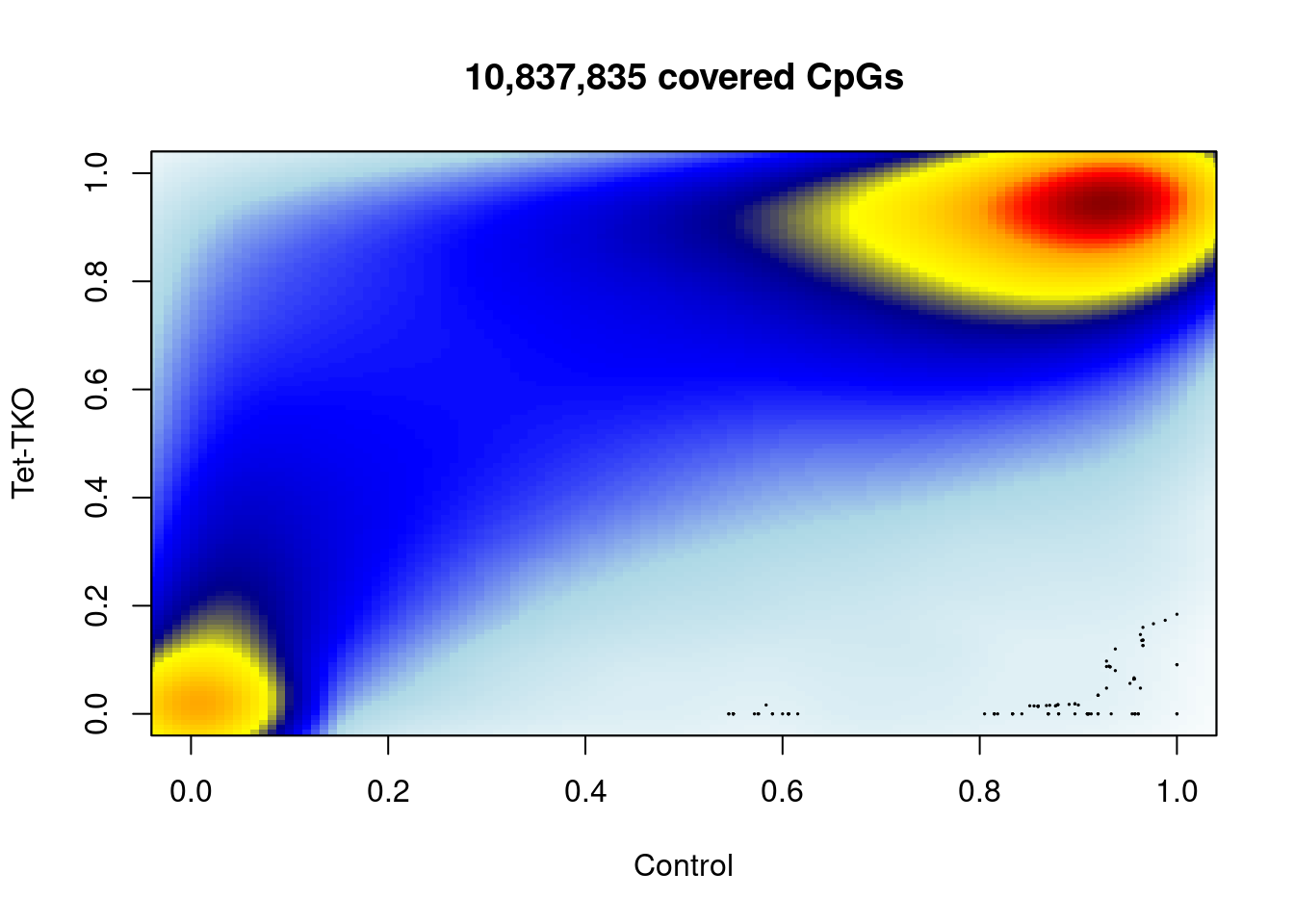
options(repr.plot.width = 5, repr.plot.height = 6)
plot_smooth_legend(c("white", "lightblue", "blue", "darkblue", "yellow", "gold", "orange", "red", "darkred"))
9.5 Methylation in TADs
We took TAD defnitions from Nagano et al., 2017:
tad_intervs <- gintervals.load("scell.nextera.pool_good_hyb_es_b_TADs") %>% as_tibble()
dim(tad_intervs)## [1] 2461 4We can then extract average methylation in TADs and then remove from it (‘punctuate’) the methylation calls that are at k4me3 or k27me3 peaks, or that are at methylation dips or ATAC peaks:
tads_meth <- get_cpg_meth_tads(tad_intervs, meth_dips, k4me3_peaks, k27me3_peaks, atac_peaks) %cache_df% here("output/tads_meth.tsv") %>% as_tibble()9.5.1 Compare TAD methylation in Ctrl and TET-tko (Figure 5C)
options(repr.plot.width = 7, repr.plot.height = 7)
df <- tads_meth %>%
filter(ctrl.cov >= 3e3, tet_tko.cov >= 3e3) %>%
mutate(l = end - start) %>%
filter(l >= 5e4) %>%
mutate(ctrl_bins = cut(ctrl, c(0.7, 0.8, 0.83, 0.86, 0.89, 1))) %>%
select(chrom, start, end, ctrl_bins, Ctrl = ctrl, TKO = tet_tko) %>%
gather("type", "meth", -(chrom:ctrl_bins))
df %>% count(ctrl_bins)## # A tibble: 5 x 2
## ctrl_bins n
## 1 (0.7,0.8] 38
## 2 (0.8,0.83] 176
## 3 (0.83,0.86] 864
## 4 (0.86,0.89] 2746
## 5 (0.89,1] 866p_tads_boxp <- df %>%
ggplot(aes(x = ctrl_bins, y = meth, fill = type)) +
geom_boxplot(lwd = 0.01, outlier.size = 0.001, fatten = 0.1) +
scale_fill_manual(name = "", values = c("Ctrl" = "#00468BFF", "TKO" = "#ED0000FF")) +
theme(aspect.ratio = 1) +
vertical_labs() +
xlab("Ctrl meth.") +
ylab("TADs meth.")
p_tads_boxp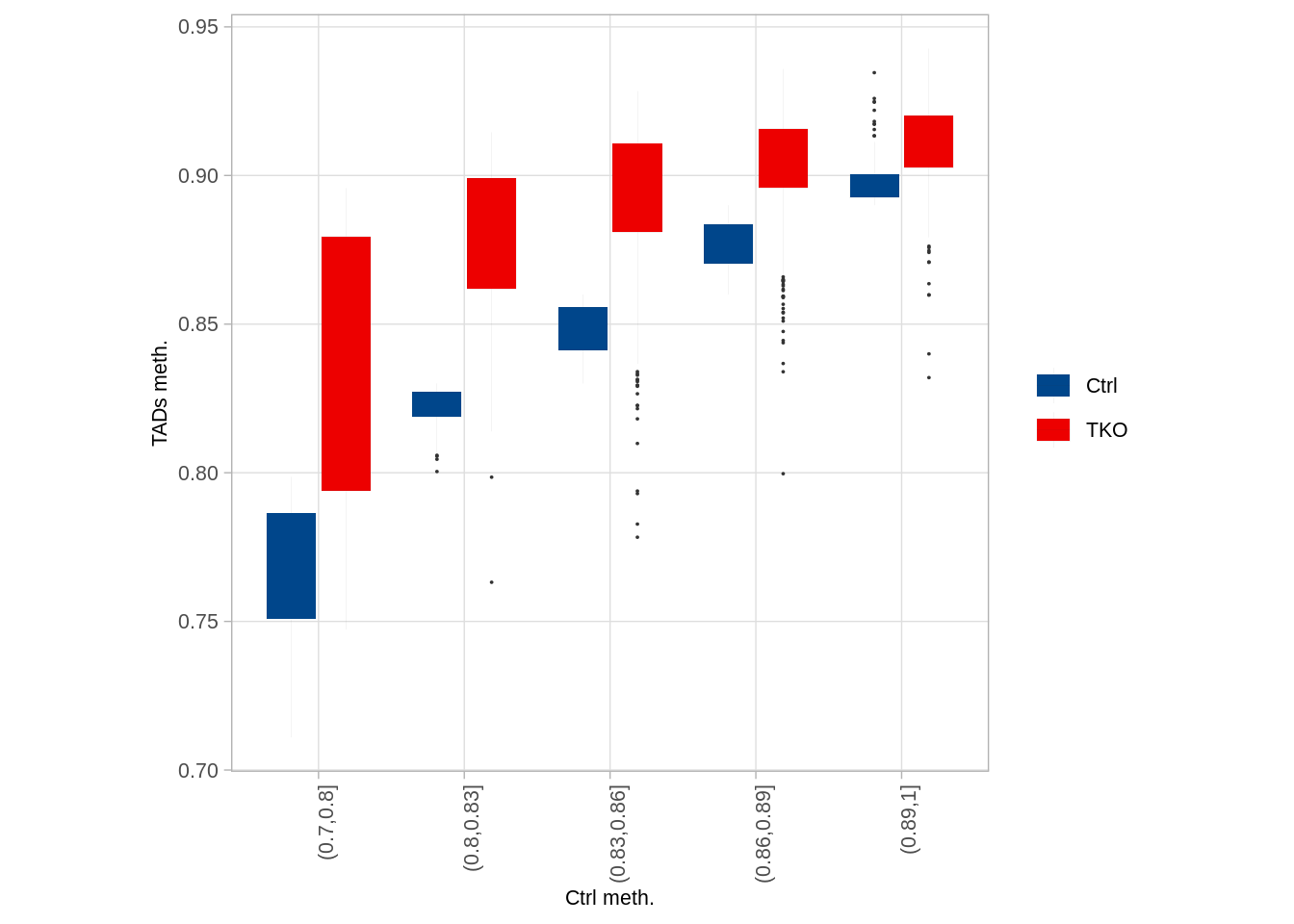
df <- tads_meth %>%
filter(ctrl.cov >= 3e3, tet_tko.cov >= 3e3) %>%
mutate(l = end - start) %>%
filter(l >= 5e4) %>%
summarise(
tet_tko_mean = mean(tet_tko),
tet_tko_sd = sd(tet_tko, na.rm = TRUE),
ctrl_mean = mean(ctrl),
ctrl_sd = sd(ctrl, na.rm = TRUE),
e7.5_mean = mean(e7.5),
e7.5_sd = sd(e7.5, na.rm = TRUE),
e8.5_mean = mean(e8.5),
e8.5_sd = sd(e8.5, na.rm = TRUE)
)
df## # A tibble: 1 x 8
## tet_tko_mean tet_tko_sd ctrl_mean ctrl_sd e7.5_mean e7.5_sd e8.5_mean
## 1 0.9017096 0.02115242 0.8723345 0.02194502 0.7953908 0.03149332 0.8195952
## e8.5_sd
## 1 0.02755257## # A tibble: 1 x 8
## tet_tko_mean tet_tko_sd ctrl_mean ctrl_sd e7.5_mean e7.5_sd e8.5_mean e8.5_sd
## 1 0.9 0.02 0.87 0.02 0.8 0.03 0.82 0.03## Warning in ks.test(tads_meth$ctrl, tads_meth$tet_tko): p-value will be
## approximate in the presence of ties##
## Two-sample Kolmogorov-Smirnov test
##
## data: tads_meth$ctrl and tads_meth$tet_tko
## D = 0.62739, p-value < 2.2e-16
## alternative hypothesis: two-sidedWe can see that that the variation in background methylation between TADs was greatly diminished in Tet-TKO cells.
9.5.2 Figure S6A
options(repr.plot.width = 7, repr.plot.height = 7)
df <- tads_meth %>%
filter(ctrl.cov >= 3e3, tet_tko.cov >= 3e3) %>%
mutate(l = end - start) %>%
filter(l >= 5e4) %>%
mutate(ctrl_bins = cut(ctrl, c(0.7, 0.8, 0.83, 0.86, 0.89, 1))) %>%
select(chrom, start, end, ctrl_bins, Ctrl = ctrl, WT = e8.5) %>%
gather("type", "meth", -(chrom:ctrl_bins))
df %>% count(ctrl_bins)## # A tibble: 5 x 2
## ctrl_bins n
## 1 (0.7,0.8] 38
## 2 (0.8,0.83] 176
## 3 (0.83,0.86] 864
## 4 (0.86,0.89] 2746
## 5 (0.89,1] 866p_tads_wt_boxp <- df %>%
ggplot(aes(x = ctrl_bins, y = meth, fill = type)) +
geom_boxplot(lwd = 0.01, outlier.size = 0.001, fatten = 0.1) +
scale_fill_manual(name = "", values = c("Ctrl" = "#00468BFF", "WT" = "#42B540FF")) +
theme(aspect.ratio = 1) +
vertical_labs() +
xlab("Ctrl meth.") +
ylab("TADs meth.")
p_tads_wt_boxp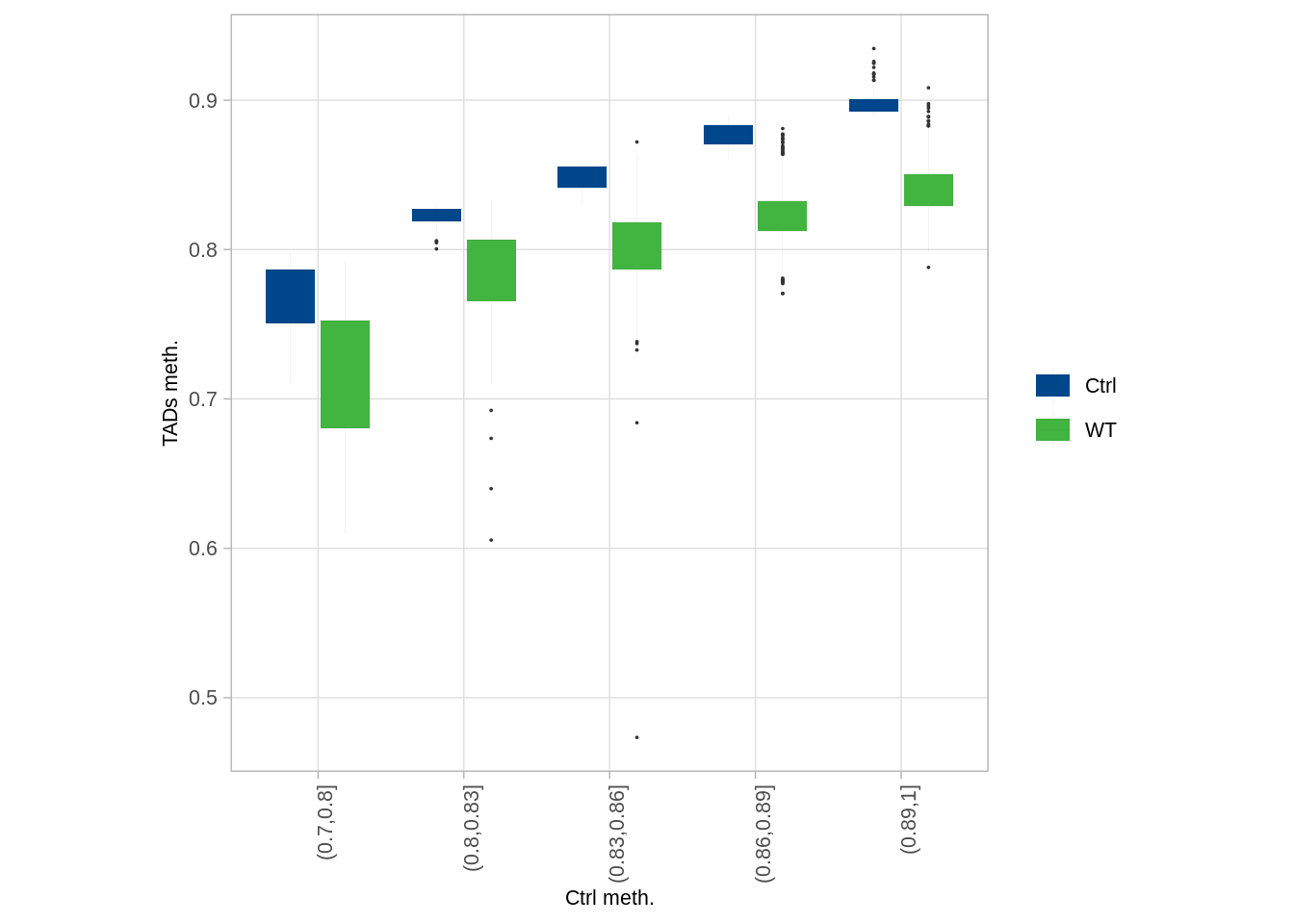
We can see that our analysis infers higher methylation levels in the embryo compared to some previously published WT data. This is likely due to the elimination of extraembryonic ectoderm from the analysis, which represents a more hypomethylated (Smith et al., 2017) cell population that can affect the estimation of average methylation when not excluded.
9.6 TAD methylation at different TOR regimes (Figure 5D)
options(repr.plot.width = 7, repr.plot.height = 7)
p_tads_tor <- tads_meth %>%
filter(ctrl.cov >= 3e3, tet_tko.cov >= 3e3) %>%
mutate(l = end - start) %>%
filter(l >= 5e4) %>%
rename(Ctrl = ctrl, TKO = tet_tko) %>%
pivot_longer(Ctrl:TKO, names_to = "type", values_to = "meth") %>%
ggplot(aes(x = -tor, y = meth, color = type)) +
geom_smooth(method = lm, se = FALSE) +
geom_point(size = 0.01, alpha = 0.1) +
theme(aspect.ratio = 1) +
xlab("TOR") +
ylab("Methylation") +
scale_color_manual(name = "", values = c("Ctrl" = "#00468BFF", "TKO" = "#ED0000FF")) +
scale_x_continuous(breaks = c(-1, 1), labels = c("Early", "Late")) +
vertical_labs()
p_tads_tor## `geom_smooth()` using formula 'y ~ x'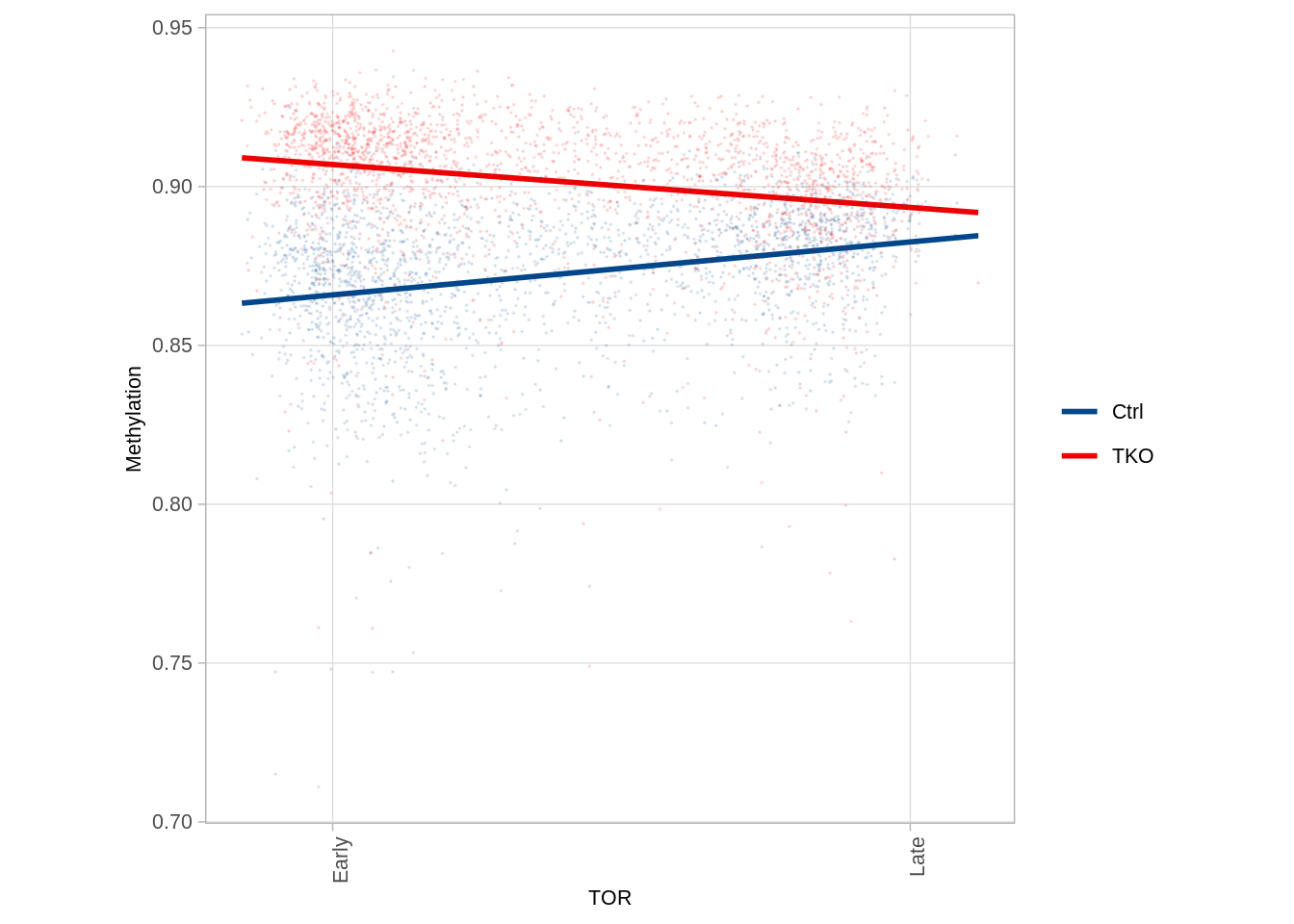
tads_meth %>%
filter(ctrl.cov >= 3e3, tet_tko.cov >= 3e3) %>%
mutate(l = end - start) %>%
filter(l >= 5e4) %>%
pivot_longer(ctrl:tet_tko, names_to = "type", values_to = "meth") %>%
group_by(type) %>%
summarise(cor = cor(meth, tor))## # A tibble: 2 x 2
## type cor
## 1 ctrl -0.2692618
## 2 tet_tko 0.2265352We see that that lower background methylation is linked with early replicating TADs in control and a reciprocal effect in Tet-TKO. These data support a role for widespread TET-mediated de-methylation in early replicating TADs, which is lost upon Tet knockout. Such de-methylation may rely on the enhanced accessibility of early replicating domains as part of the chromosomal A-compartment (López-Moyado et al., 2019; Pope et al., 2014).
9.7 Low methylation hotspots
We extract chromatin marks at hotspots of low methylation (control methylation <0.25):
define_chip_vtracks()
low_meth_df <- meth_df_comp %>%
select(chrom, start, end, ctrl, tet_tko, e8.5, ctrl.cov, tet_tko.cov, e8.5.cov) %>%
gextract.left_join(
c(
"pmax(-log2(1-ecto_k27me3),-log2(1-endo_k27me3),-log2(1-meso_k27me3),-log2(1-ps_k27me3),-log2(1-e6_epi_k27me3),-log2(1-e5_epi_k27me3),-log2(1-e6_ve_k27me3),-log2(1-e10_fc_k27me3),-log2(1-e10_forebrain_k27me3),-log2(1-e10_heart_k27me3),-log2(1-e10_hindbrain_k27me3),-log2(1-e10_limb_k27me3),-log2(1-e10_midbrain_k27me3),-log2(1-es_k27me3), na.rm=TRUE)",
"pmax(-log2(1-e10_limb_k4me3),-log2(1-e10_heart_k4me3),-log2(1-e10_midbrain_k4me3),-log2(1-e10_hindbrain_k4me3),-log2(1-e10_forebrain_k4me3),-log2(1-e11_liver_k4me3),-log2(1-ecto_k4me3),-log2(1-endo_k4me3),-log2(1-meso_k4me3),-log2(1-ps_k4me3),-log2(1-e6_epi_k4me3),-log2(1-e5_epi_k4me3),-log2(1-e6_ve_k4me3), na.rm=TRUE)"
),
iterator = ., intervals = ., colnames = c("k27me3", "k4me3")
) %>%
select(-(chrom1:end1)) %cache_df%
here("output/meth_marks_smoothed.tsv") %>%
as_tibble()9.7.1 Figure S6B
options(repr.plot.width = 14, repr.plot.height = 7)
df <- low_meth_df %>%
filter(ctrl.cov >= 10) %>%
mutate(low_meth = ifelse(ctrl <= 0.25, "Low meth.", "Other"))
p_k4me3 <- df %>%
ggplot(aes(x = k4me3, color = low_meth, y = 1 - ..y..)) +
stat_ecdf() +
scale_color_manual("", values = c("darkblue", "darkgray")) +
scale_y_continuous(labels = scales::percent) +
ylab("% of loci") +
xlab("H3K4me3")
p_k27me3 <- df %>%
ggplot(aes(x = k27me3, color = low_meth, y = 1 - ..y..)) +
stat_ecdf() +
scale_color_manual("", values = c("darkblue", "darkgray")) +
scale_y_continuous(labels = scales::percent) +
ylab("% of loci") +
xlab("H3K27me3")
p_k4me3 + p_k27me3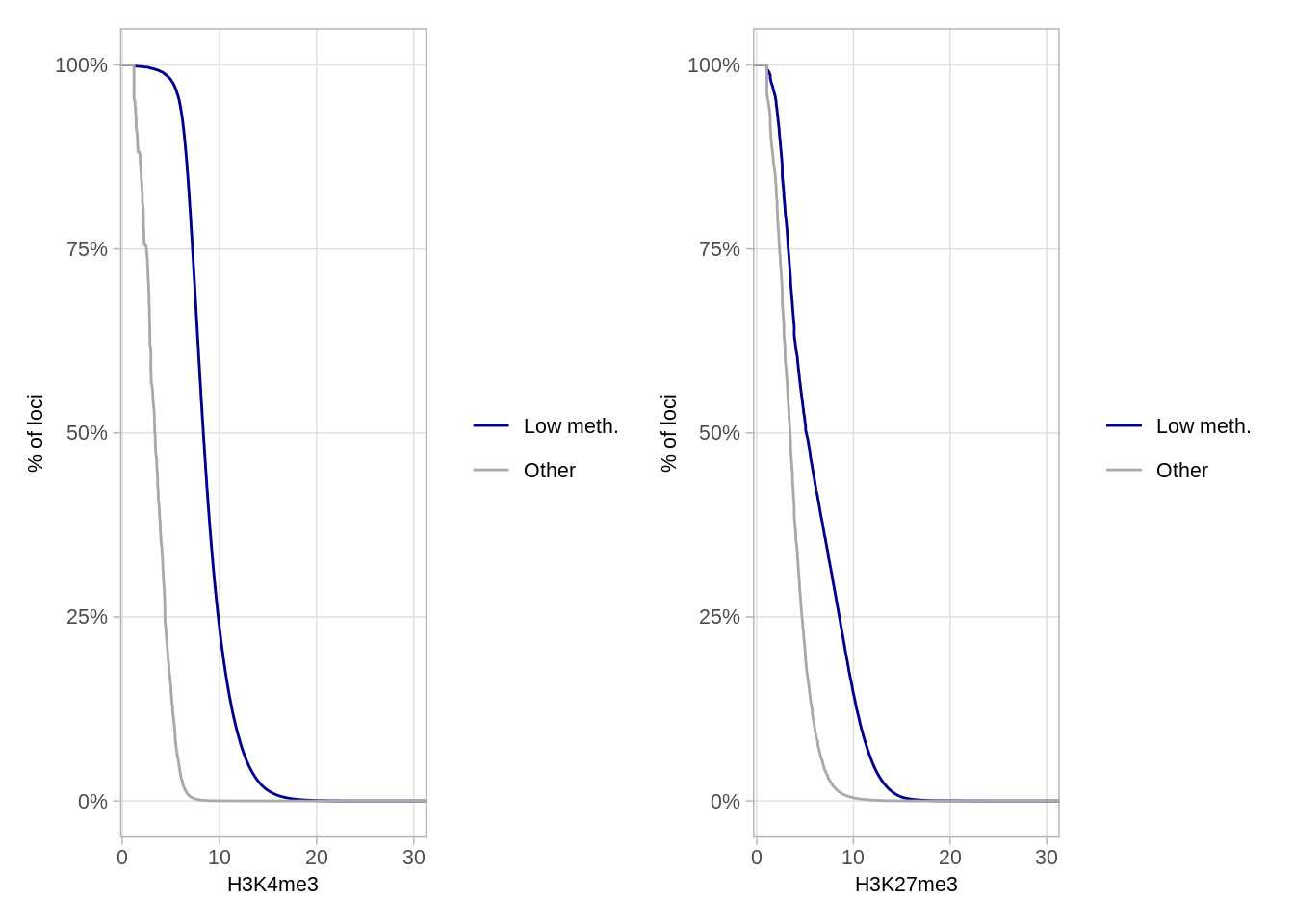
9.8 K4me3/K27me3 hospots (Figure 5E)
9.8.1 K4me3 hotspots
d_k4me3 <- extract_meth_at_context(k4me3_peaks) %cache_df% here("output/k4me3_meth_comp.tsv") %>% as_tibble()options(repr.plot.width = 7, repr.plot.height = 7)
df_k4me3 <- d_k4me3 %>%
filter(ctrl.cov >= 50, tet_tko.cov >= 50) %>%
gintervals.neighbors1(k27me3_peaks) %>%
mutate(type = ifelse(dist == 0, "bivalent", "other")) %>%
select(-(chrom1:dist)) %>%
mutate(l = end - start) %>%
filter(abs(l) >= 3e3)
p_k4me3 <- df_k4me3 %>%
ggplot(aes(x = ctrl, y = tet_tko, color = type)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
scale_color_manual(name = "", values = c("bivalent" = "darkred", "other" = "darkgray")) +
ggtitle("K4me3") +
xlab("WT") +
ylab("TKO ") +
xlim(0, 1) +
ylim(0, 1)
p_k4me3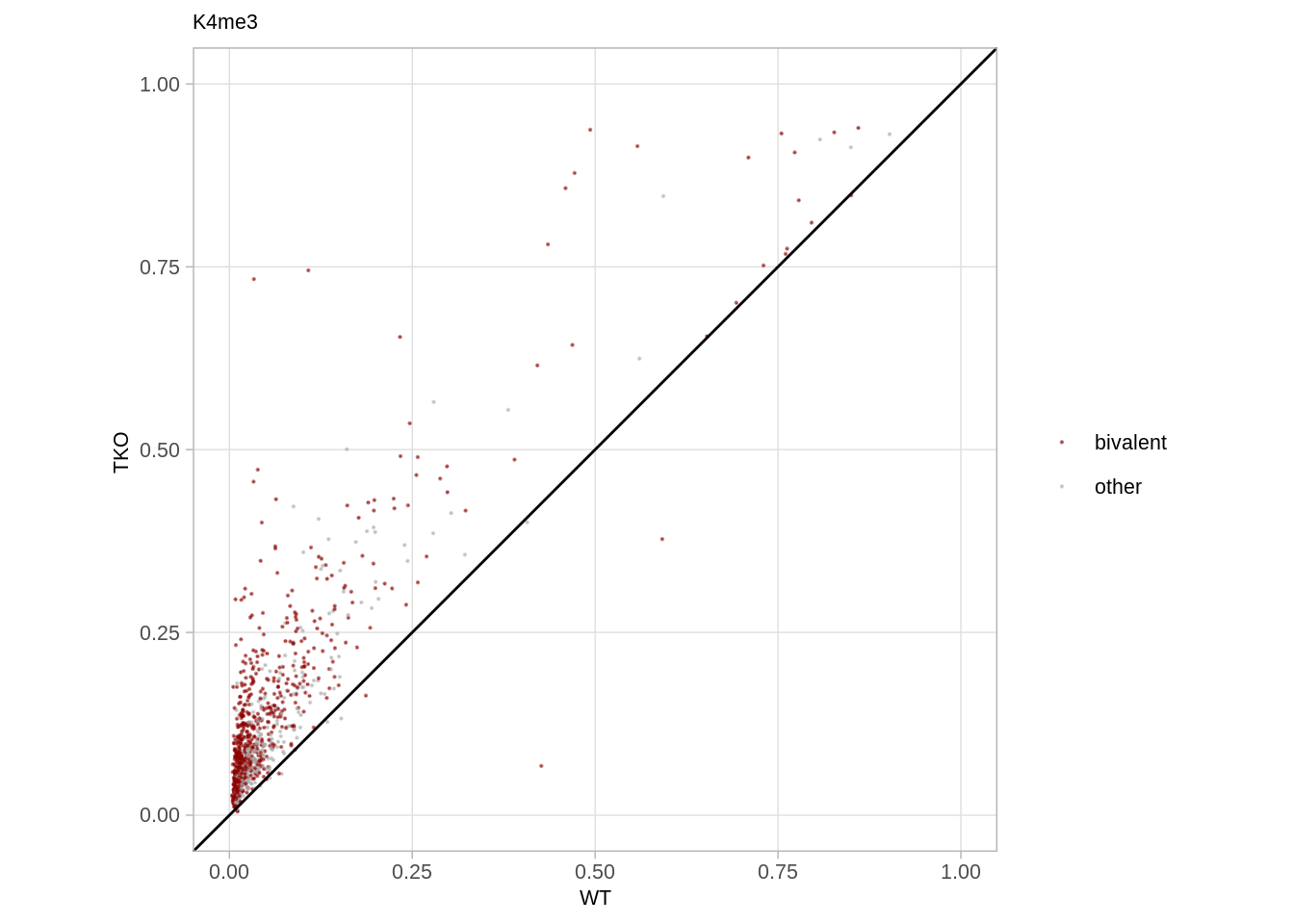
## # A tibble: 2 x 2
## type n
## 1 bivalent 668
## 2 other 285## [1] 953df_k4me3 %>%
select(chrom:end, ctrl, tet_tko, type) %>%
mutate(diff = tet_tko - ctrl) %>%
filter(diff >= 0) %>%
summarise(min_diff = min(diff), max_diff = max(diff), m = mean(diff), sd = sd(diff)) %>%
summarise_at(vars(everything()), scales::percent)## # A tibble: 1 x 4
## min_diff max_diff m sd
## 1 0% 70% 8% 7%9.8.2 K27me3 hotspots
d_k27me3 <- extract_meth_at_context(k27me3_peaks) %cache_df% here("output/k27me3_meth_comp.tsv") %>% as_tibble()Polycomb without methylation of k4me3 regions (“punctuated”):
d_k27me3_punc <- gextract_meth(c(comb_tracks, "e8_5", "Zhang_Nature_Genetics_2017.Ect_mCG", "Zhang_Nature_Genetics_2017.End_mCG", "Zhang_Nature_Genetics_2017.Mes_mCG", "Zhang_Nature_Genetics_2017.E65Epi_mCG"), names = c(comb_names, "e8.5", "ecto", "endo", "meso", "epi"), extract_meth_calls = TRUE, intervals = k27me3_peaks, join_intervals = TRUE) %>%
mutate(e7.5.meth = ecto.meth + endo.meth + meso.meth, e7.5.cov = ecto.cov + endo.cov + meso.cov, e7.5 = e7.5.meth / e7.5.cov) %>%
gintervals.neighbors1(k4me3_peaks) %>%
filter(dist != 0) %>%
group_by(chrom1, start1, end1) %>%
summarise_at(vars(ends_with("cov"), ends_with("meth")), sum, na.rm = TRUE) %>%
rename(chrom = chrom1, start = start1, end = end1) %>%
mutate(ctrl = ctrl.meth / ctrl.cov, tet_tko = tet_tko.meth / tet_tko.cov, e8.5 = e8.5.meth / e8.5.cov, e7.5 = e7.5.meth / e7.5.cov, epi = epi.meth / epi.cov) %>%
ungroup() %cache_df% here("output/k27me3_meth_comp_k4me3_punc.tsv") %>%
as_tibble()options(repr.plot.width = 7, repr.plot.height = 7)
df <- d_k27me3 %>%
filter(ctrl.cov >= 50, tet_tko.cov >= 50) %>%
gintervals.neighbors1(k4me3_peaks) %>%
mutate(type = ifelse(dist == 0, "bivalent", "other")) %>%
select(-(chrom1:dist)) %>%
mutate(l = end - start) %>%
filter(abs(l) >= 3e3)
p_k27me3 <- df %>%
ggplot(aes(x = ctrl, y = tet_tko, color = type)) +
geom_point(size = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme_bw() +
theme(aspect.ratio = 1) +
scale_color_manual(name = "", values = c("bivalent" = "darkred", "other" = "darkgray")) +
ggtitle("H3K27me3") +
xlab("Ctrl") +
ylab("TKO")
p_k27me3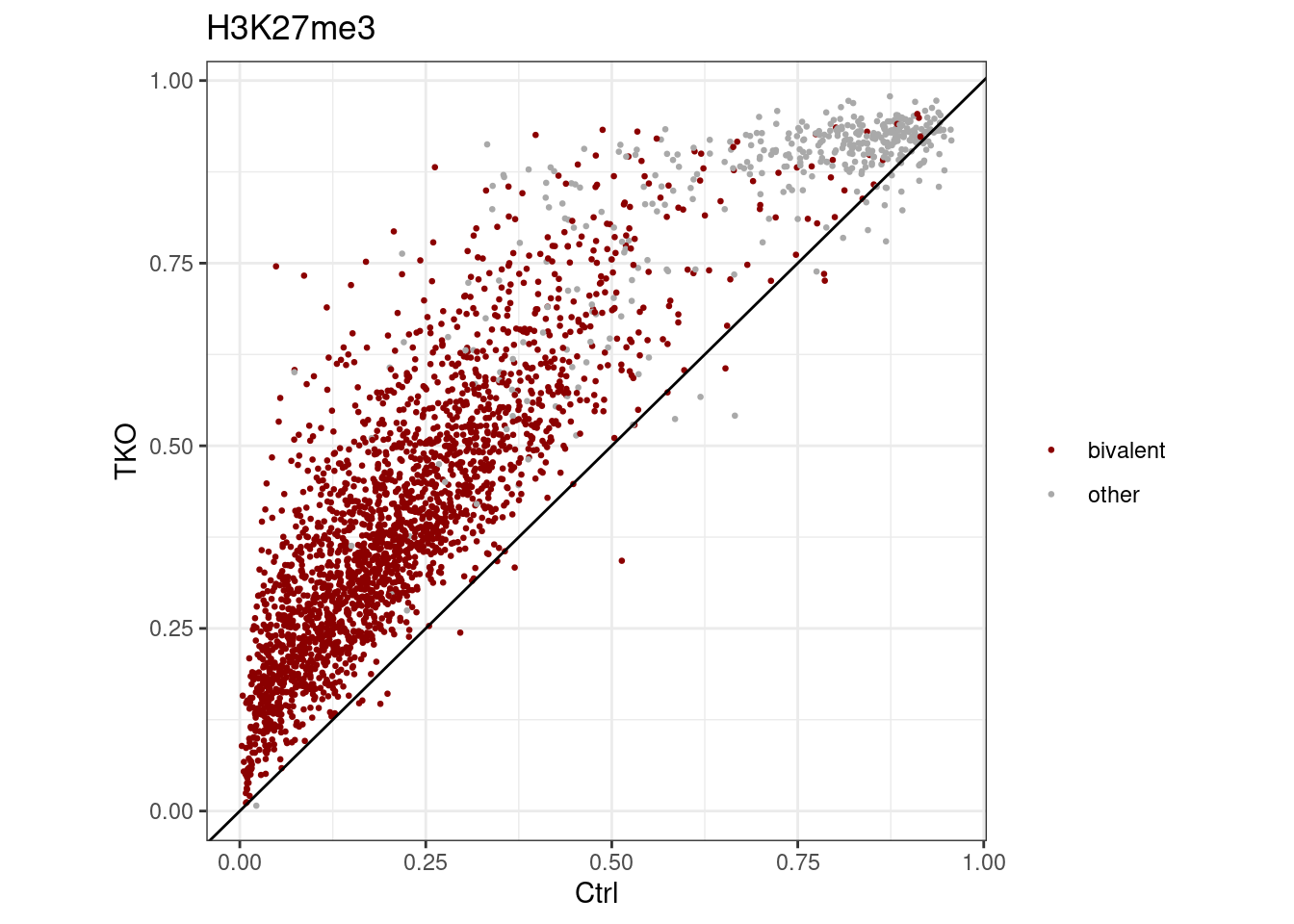
## # A tibble: 2 x 2
## type n
## 1 bivalent 2298
## 2 other 402## [1] 2700H3k27me3 hotspots while removing H3K4me3 methylation:
options(repr.plot.width = 7, repr.plot.height = 7)
df_punc <- d_k27me3_punc %>%
filter(ctrl.cov >= 50, tet_tko.cov >= 50) %>%
gintervals.neighbors1(k4me3_peaks) %>%
mutate(type = ifelse(dist == 0, "bivalent", "other")) %>%
select(-(chrom1:dist)) %>%
mutate(l = end - start) %>%
filter(abs(l) >= 3e3)
p_k27_punc <- df_punc %>%
ggplot(aes(x = ctrl, y = tet_tko, color = type)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
scale_color_manual(name = "", values = c("bivalent" = "darkred", "other" = "darkgray")) +
ggtitle("H3K27me3") +
xlab("Ctrl") +
ylab("TKO")
p_k27_punc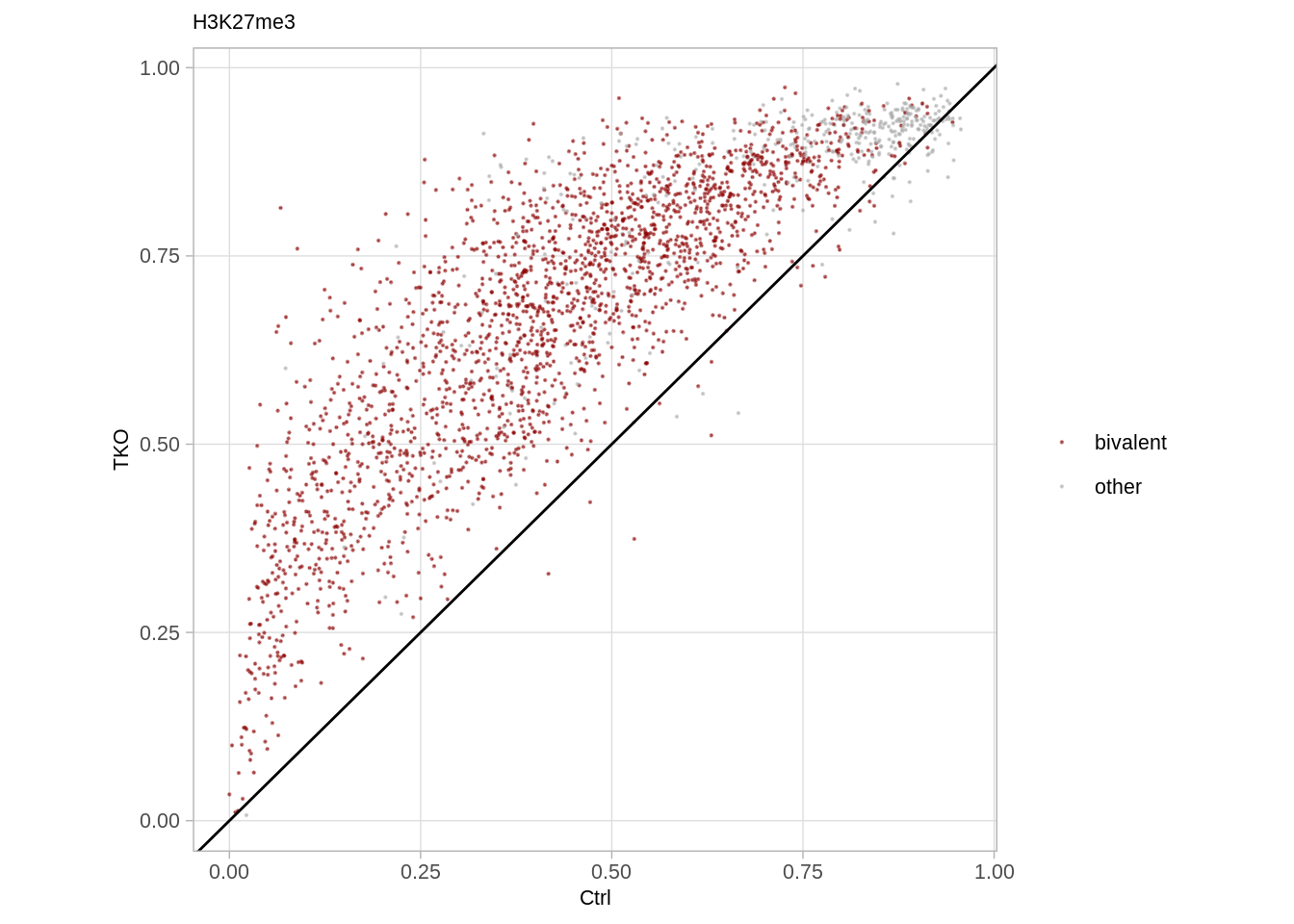
df_punc %>%
select(chrom:end, ctrl, tet_tko) %>%
mutate(diff = tet_tko - ctrl) %>%
filter(diff >= 0) %>%
summarise(min_diff = min(diff), max_diff = max(diff), m = mean(diff), sd = sd(diff)) %>%
summarise_at(vars(everything()), scales::percent)## # A tibble: 1 x 4
## min_diff max_diff m sd
## 1 0% 75% 23% 12%bind_rows(df_k4me3, df_punc) %>%
select(chrom:end, ctrl, tet_tko, type) %>%
mutate(diff = tet_tko - ctrl) %>%
filter(diff >= 0) %>%
summarise(min_diff = min(diff), max_diff = max(diff), m = mean(diff), sd = sd(diff)) %>%
summarise_at(vars(everything()), scales::percent)## # A tibble: 1 x 4
## min_diff max_diff m sd
## 1 0% 75% 19% 13%n_above <- bind_rows(df_k4me3, df_punc) %>%
select(chrom:end, ctrl, tet_tko, type) %>%
filter(ctrl <= 0.25, tet_tko >= 0.5) %>%
nrow()
scales::percent(n_above / nrow(bind_rows(df_k4me3, df_punc)))## [1] "5%"## # A tibble: 2 x 2
## type n
## 1 bivalent 2185
## 2 other 402## [1] 25879.9 Loci linked with CTCF occupancy (Figure 5F)
tet_df <- meth_df_comp %>% filter(ctcf >= 0.999)
tet_df <- tet_df %>%
gintervals.neighbors1(k4me3_peaks) %>%
mutate(type = ifelse(dist == 0, "k4me3", "other")) %>%
select(chrom:end, ctrl.cov, tet_tko.cov, ctrl, tet_tko, ctcf, type)options(repr.plot.width = 7, repr.plot.height = 7)
min_cov <- 20
p_ctcf <- tet_df %>%
filter(ctrl.cov >= min_cov, tet_tko.cov >= min_cov) %>%
ggplot(aes(x = ctrl, y = tet_tko)) +
geom_point(size = 0.001, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline(color = "red") +
facet_wrap(~type) +
ggtitle("CTCF peaks") +
xlab("WT") +
ylab("TKO")
p_ctcf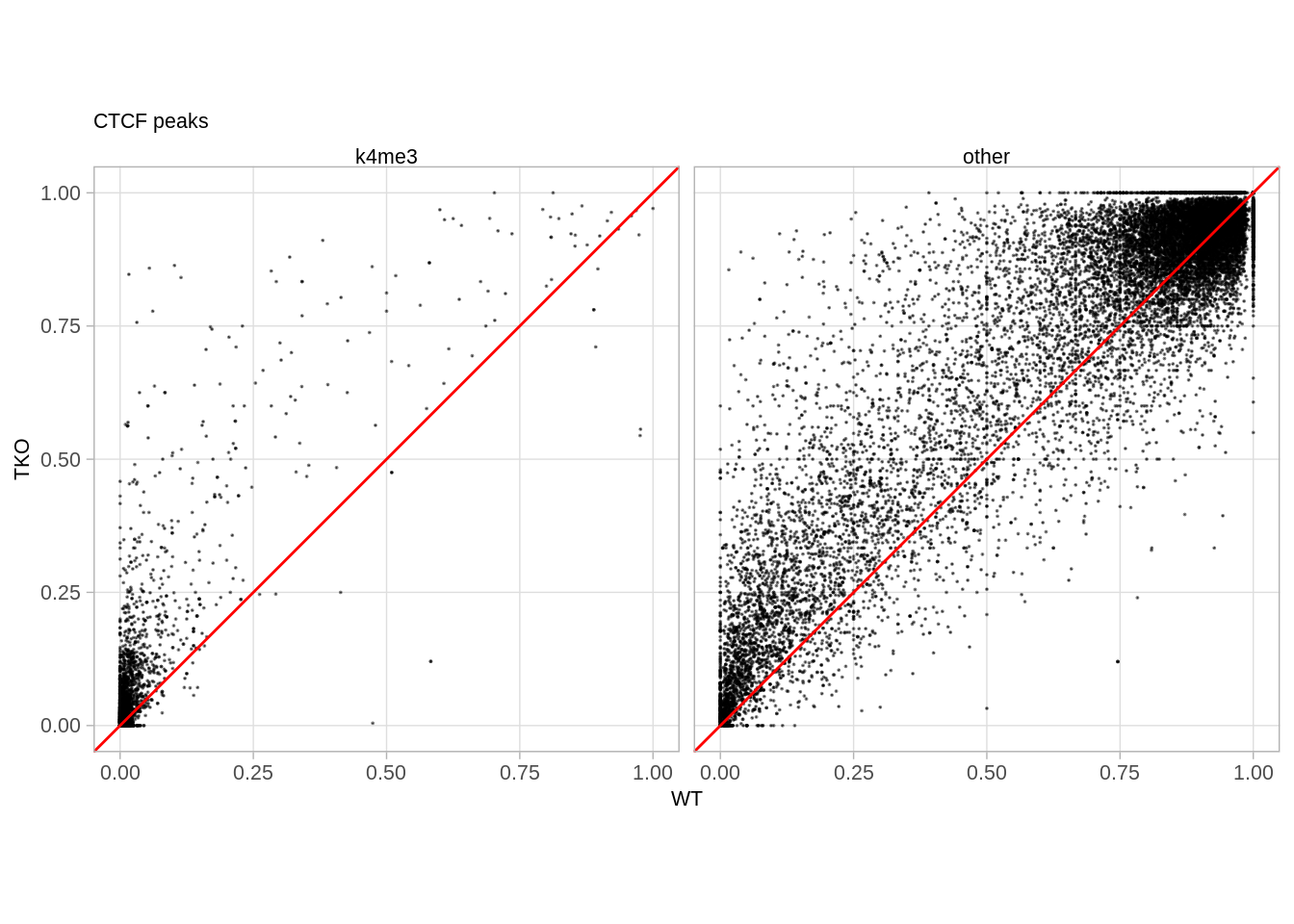
## # A tibble: 2 x 2
## type n
## 1 k4me3 3276
## 2 other 178609.10 Methylation at Exons and Non-H3K4me3 Promoters (Figure 5G)
9.10.1 Exons
d_exons <- extract_meth_at_context("intervs.global.exons") %cache_df% here("output/exons_meth_comp.tsv") %>% as_tibble()
d_exons <- d_exons %>%
gintervals.neighbors1(k4me3_peaks) %>%
filter(abs(dist) > 0) %>%
select(-(chrom1:dist)) %>%
gintervals.neighbors1(k27me3_peaks) %>%
filter(abs(dist) > 0) %>%
select(-(chrom1:dist))options(repr.plot.width = 7, repr.plot.height = 7)
p_exons <- d_exons %>%
filter(ctrl.cov >= 30, tet_tko.cov >= 30) %>%
ggplot(aes(x = ctrl, y = tet_tko)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
ggtitle("Exons") +
xlab("Ctrl") +
ylab("TKO")
p_exons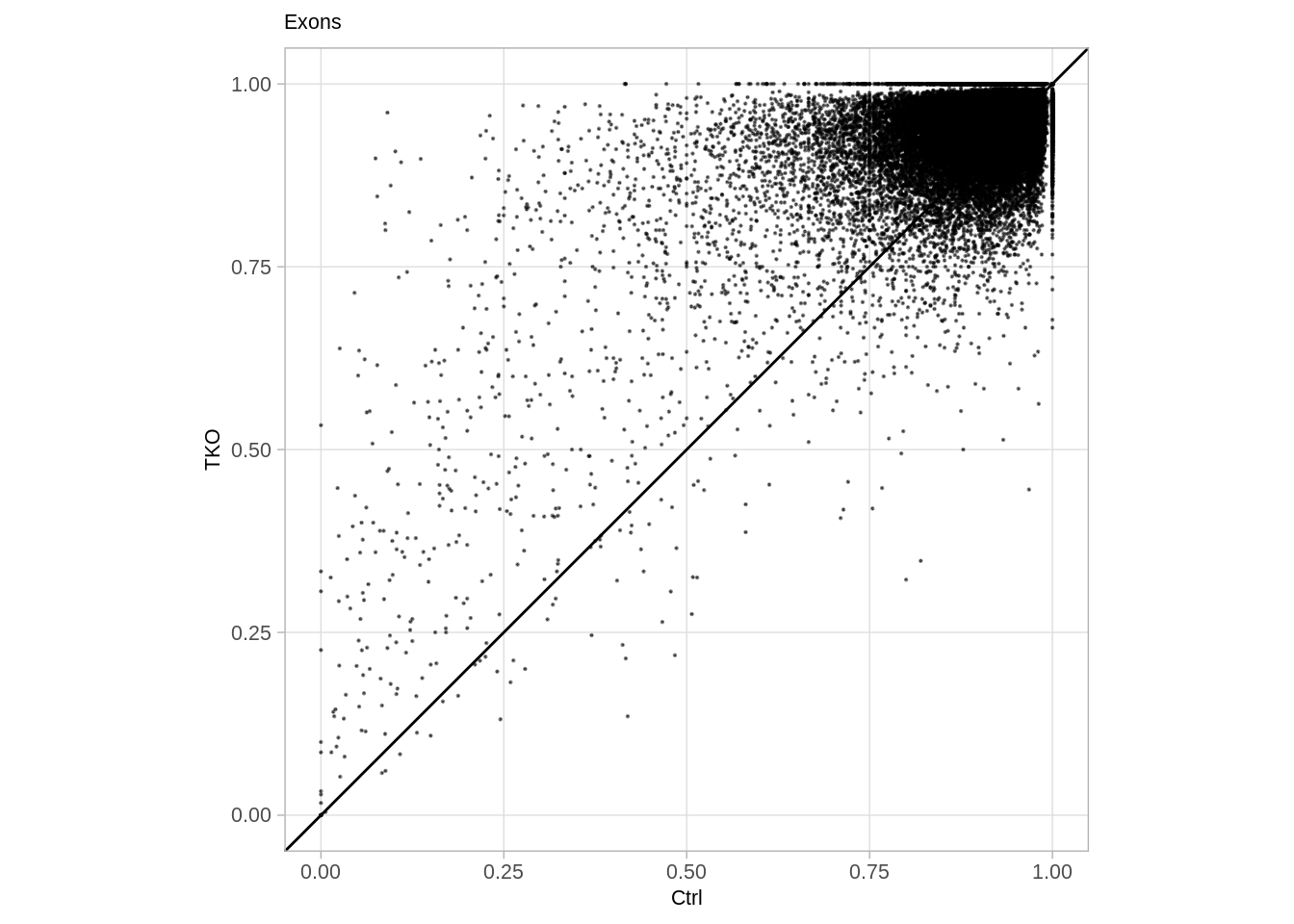
## [1] 477349.10.2 Compare with the WT (Figure S6C):
options(repr.plot.width = 7, repr.plot.height = 7)
p_exons_vs_wt <- d_exons %>%
filter(e7.5.cov >= 30, tet_tko.cov >= 30) %>%
ggplot(aes(x = e7.5, y = tet_tko)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
ggtitle("Exons") +
xlab("WT") +
ylab("TKO")
p_exons_vs_wt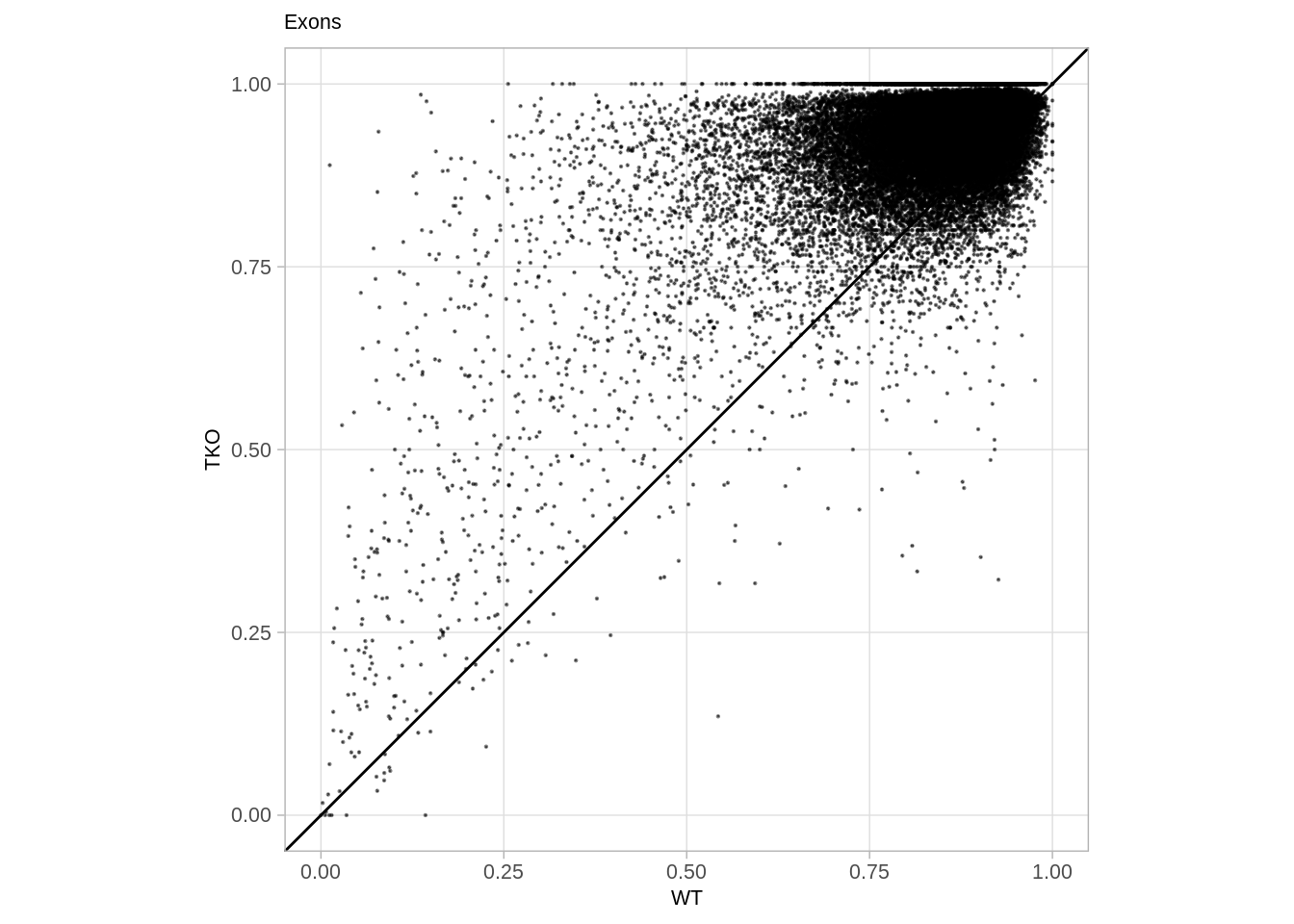
9.10.3 Promoters
d_promoters <- extract_meth_at_context(get_promoters()) %cache_df% here("output/promoter_meth_comp.tsv") %>% as_tibble()Annotate promoters to ‘k4me3’, ‘k27me3’, ‘bivalent’ and other (‘none’):
d_promoters <- d_promoters %>%
gintervals.neighbors1(k4me3_peaks) %>%
mutate(k4me3 = dist == 0) %>%
select(-(chrom1:dist)) %>%
gintervals.neighbors1(k27me3_peaks) %>%
mutate(k27me3 = dist == 0) %>%
select(-(chrom1:dist)) %>%
mutate(type = case_when(k27me3 & k4me3 ~ "bivalent", k4me3 ~ "k4me3", k27me3 ~ "k27me3", TRUE ~ "none"))Add time of replication annotation:
d_promoters <- gextract.left_join("tor", intervals = d_promoters, iterator = d_promoters) %>%
as_tibble() %>%
select(-(chrom1:end1))We match the promoters with marginal expression data from the gastrulation atlas (Mittnenzweig, Meishar et. al. Cell 2021):
atlas_egc <- readr::read_rds(here("data/atlas_egc.rds"))
atlas_marginal <- tibble(gene = rownames(atlas_egc), expr = log2(rowSums(atlas_egc) + 1e-5))9.10.3.1 Non-H3Kme3 promoters
We require a coverage of at least 30:
df <- d_promoters %>%
filter(type == "none", ctrl.cov >= 30, tet_tko.cov >= 30) %>%
left_join(get_promoters() %>% select(chrom:end, gene = geneSymbol)) %>%
left_join(atlas_marginal %>% separate_rows(gene, sep = ";")) %>%
gintervals.neighbors1(cgi_intervs) %>%
mutate(cgi = ifelse(dist == 0, "cgi", "other")) %>%
filter(!is.na(expr))## Joining, by = c("chrom", "start", "end")
## Joining, by = "gene"## [1] 3381options(repr.plot.width = 7, repr.plot.height = 7)
p_prom_none <- df %>%
ggplot(aes(x = ctrl, y = tet_tko)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
ggtitle("Non-H3K4me3 Promoters") +
xlab("Ctrl") +
ylab("TKO")
p_prom_none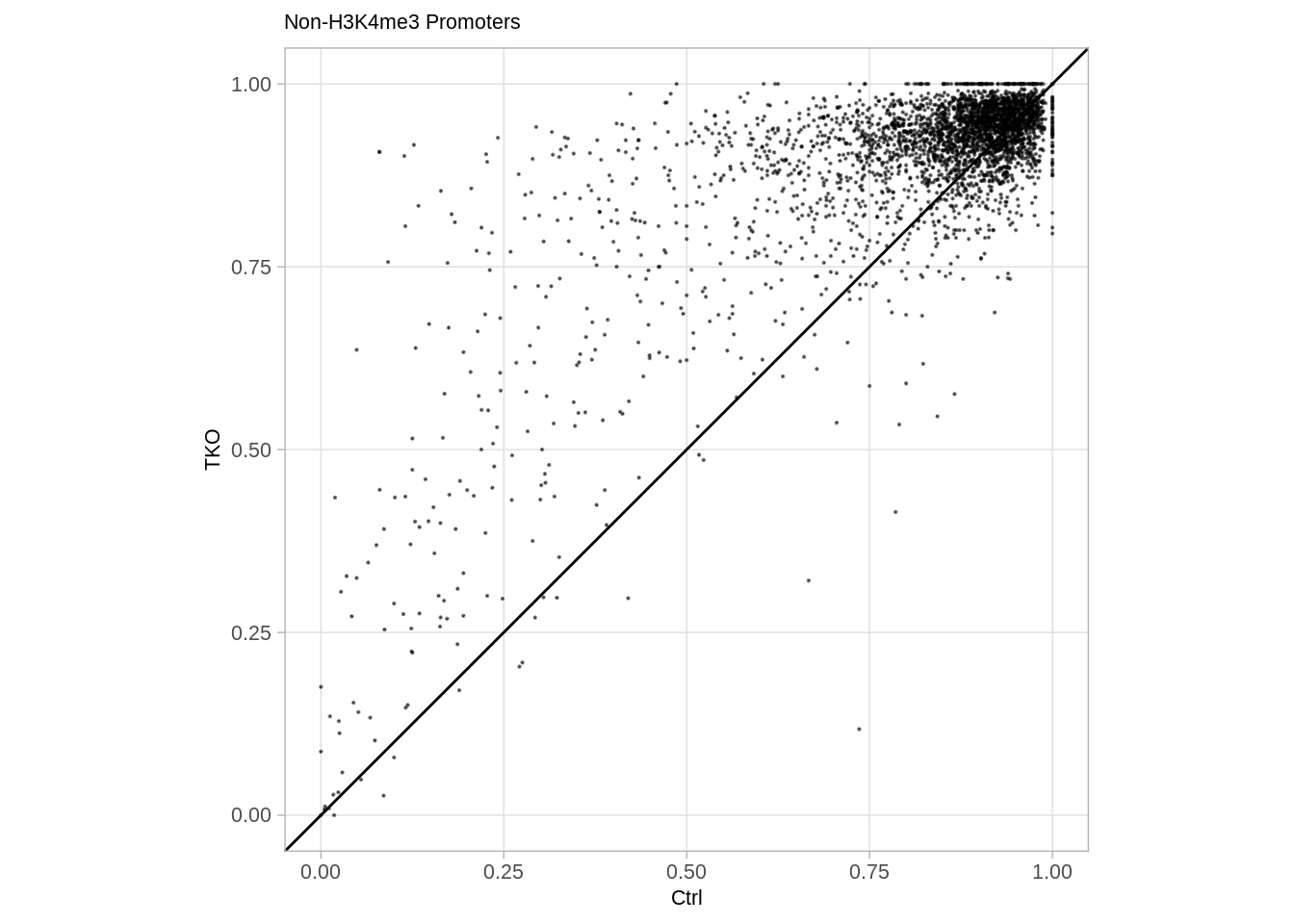
9.10.4 Compare with the WT (Figure S6C):
options(repr.plot.width = 7, repr.plot.height = 7)
p_prom_none_vs_wt <- df %>%
filter(e7.5.cov >= 30, tet_tko.cov >= 30) %>%
ggplot(aes(x = e7.5, y = tet_tko)) +
geom_point(size = 0.1, alpha = 0.5) +
theme(aspect.ratio = 1) +
geom_abline() +
theme(aspect.ratio = 1) +
ggtitle("Non-H3K4me3 Promoters") +
xlab("WT") +
ylab("TKO")
p_prom_none_vs_wt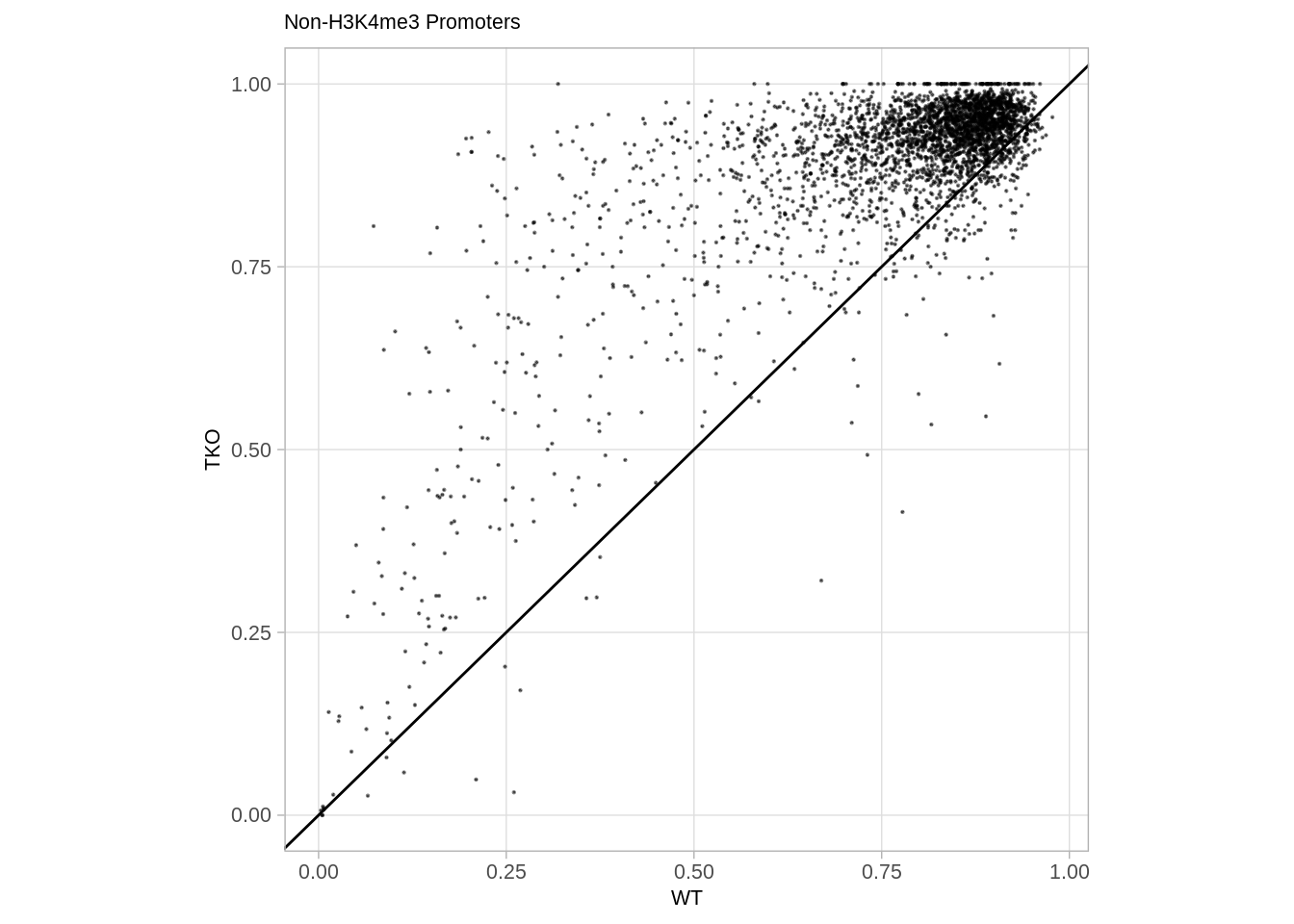
9.11 Promoter methylation vs expression (Figure S6D/S6E)
Load expression data of all the types and of epiblast:
egc_bulk <- fread(here("data/eg_bulk.tsv")) %>% as_tibble()
egc_epiblast <- fread(here("data/eg_epiblast.tsv")) %>% as_tibble()We generate a table for all the covered promoters in wt.
promoters_table <- d_promoters %>%
left_join(get_promoters() %>% select(chrom:end, strand, geneSymbol), by = c("chrom", "start", "end")) %>%
filter(ctrl.cov >= 30) %>%
separate_rows(geneSymbol, sep = ";") %>%
# when there are alternative promoters - keep the one with lower methylation
arrange(geneSymbol, ctrl) %>%
distinct(geneSymbol, .keep_all = TRUE) %>%
# add expression data
left_join(egc_bulk %>% separate_rows(gene, sep = ";") %>% rename(geneSymbol = gene), by = "geneSymbol") %>%
mutate_at(vars(ends_with("bulk")), function(x) log2(x + 1e-5)) %cache_df%
here("output/promoter_table.tsv") %>%
as_tibble()promoters_table %>%
filter(wt_bulk >= -14) %>%
count(type) %>%
mutate(p = n / sum(n), perc = scales::percent(p))## # A tibble: 4 x 4
## type n p perc
## 1 bivalent 542 0.107539683 10.8%
## 2 k27me3 21 0.004166667 0.4%
## 3 k4me3 4277 0.848611111 84.9%
## 4 none 200 0.039682540 4.0%p_expr_marks <- promoters_table %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("low", "mid", "high"))) %>%
filter(!is.na(expr)) %>%
count(expr, type) %>%
ggplot(aes(x = expr, y = n, fill = type)) +
geom_col() +
scale_fill_manual(name = "", values = c("bivalent" = "darkred", "k27me3" = "darkblue", "k4me3" = "darkgreen", "none" = "darkgray")) +
ylab("# of promoters") +
xlab("Gene expression")
p_expr_marks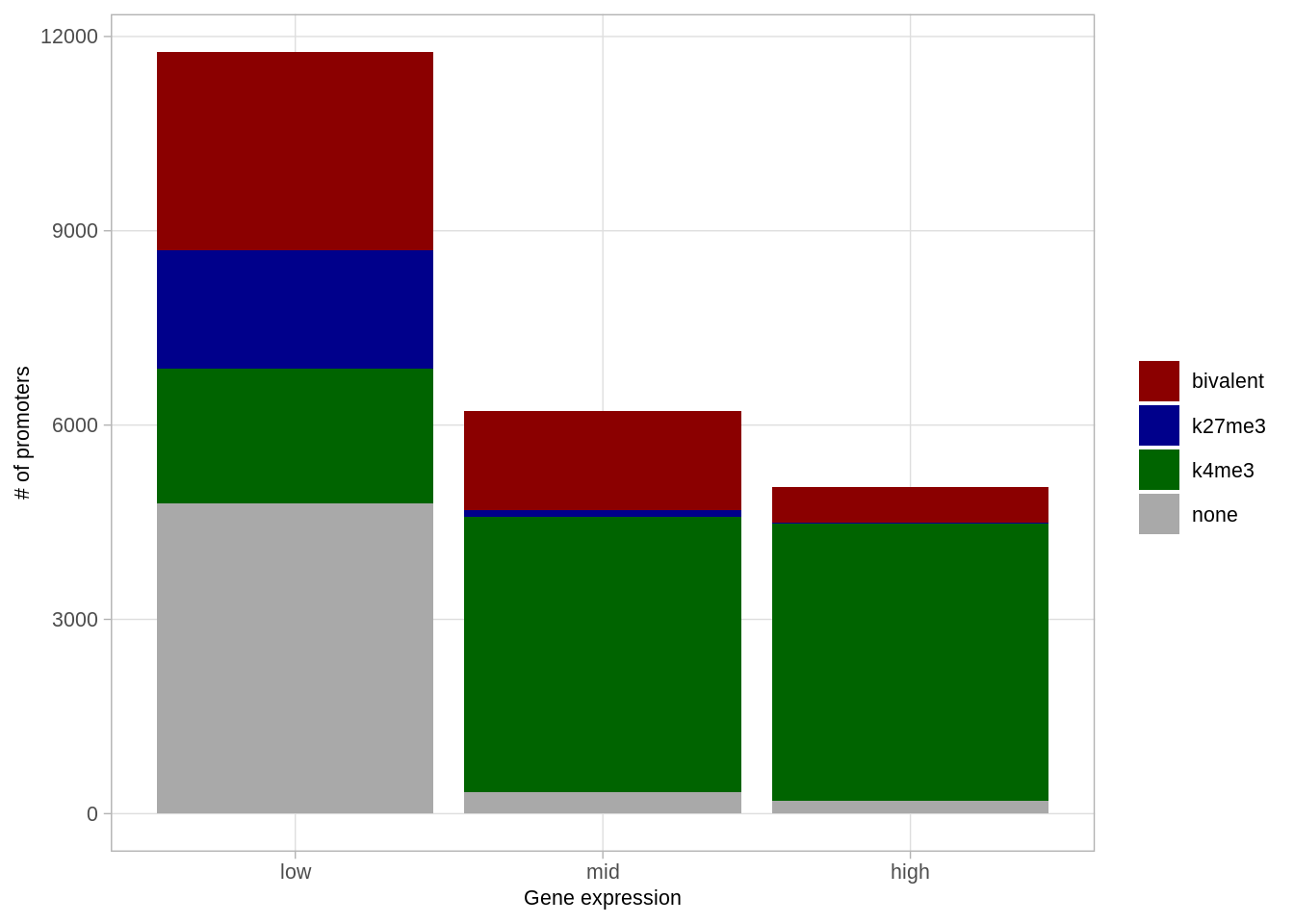
define_chip_vtracks()
intervs <- promoters_table %>% select(chrom:end, geneSymbol, wt_bulk)
marks_expr <- gextract.left_join(
c(
"pmax(-log2(1-ecto_k27me3),-log2(1-endo_k27me3),-log2(1-meso_k27me3),-log2(1-ps_k27me3),-log2(1-e6_epi_k27me3),-log2(1-e5_epi_k27me3),-log2(1-e6_ve_k27me3),-log2(1-e10_fc_k27me3),-log2(1-e10_forebrain_k27me3),-log2(1-e10_heart_k27me3),-log2(1-e10_hindbrain_k27me3),-log2(1-e10_limb_k27me3),-log2(1-e10_midbrain_k27me3),-log2(1-es_k27me3), na.rm=TRUE)",
"pmax(-log2(1-e10_limb_k4me3),-log2(1-e10_heart_k4me3),-log2(1-e10_midbrain_k4me3),-log2(1-e10_hindbrain_k4me3),-log2(1-e10_forebrain_k4me3),-log2(1-e11_liver_k4me3),-log2(1-ecto_k4me3),-log2(1-endo_k4me3),-log2(1-meso_k4me3),-log2(1-ps_k4me3),-log2(1-e6_epi_k4me3),-log2(1-e5_epi_k4me3),-log2(1-e6_ve_k4me3), na.rm=TRUE)"
),
iterator = intervs, intervals = intervs, colnames = c("k27me3", "k4me3")
) %>%
select(-(chrom1:end1)) %cache_df% here("output/marks_expr.tsv") %>%
as_tibble()options(repr.plot.width = 14, repr.plot.height = 5)
marks_expr %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("low", "mid", "high"))) %>%
filter(!is.na(expr)) %>%
ggplot(aes(x = k4me3, y = k27me3)) +
geom_point(size = 0.1) +
facet_wrap(~expr) +
theme(aspect.ratio = 1)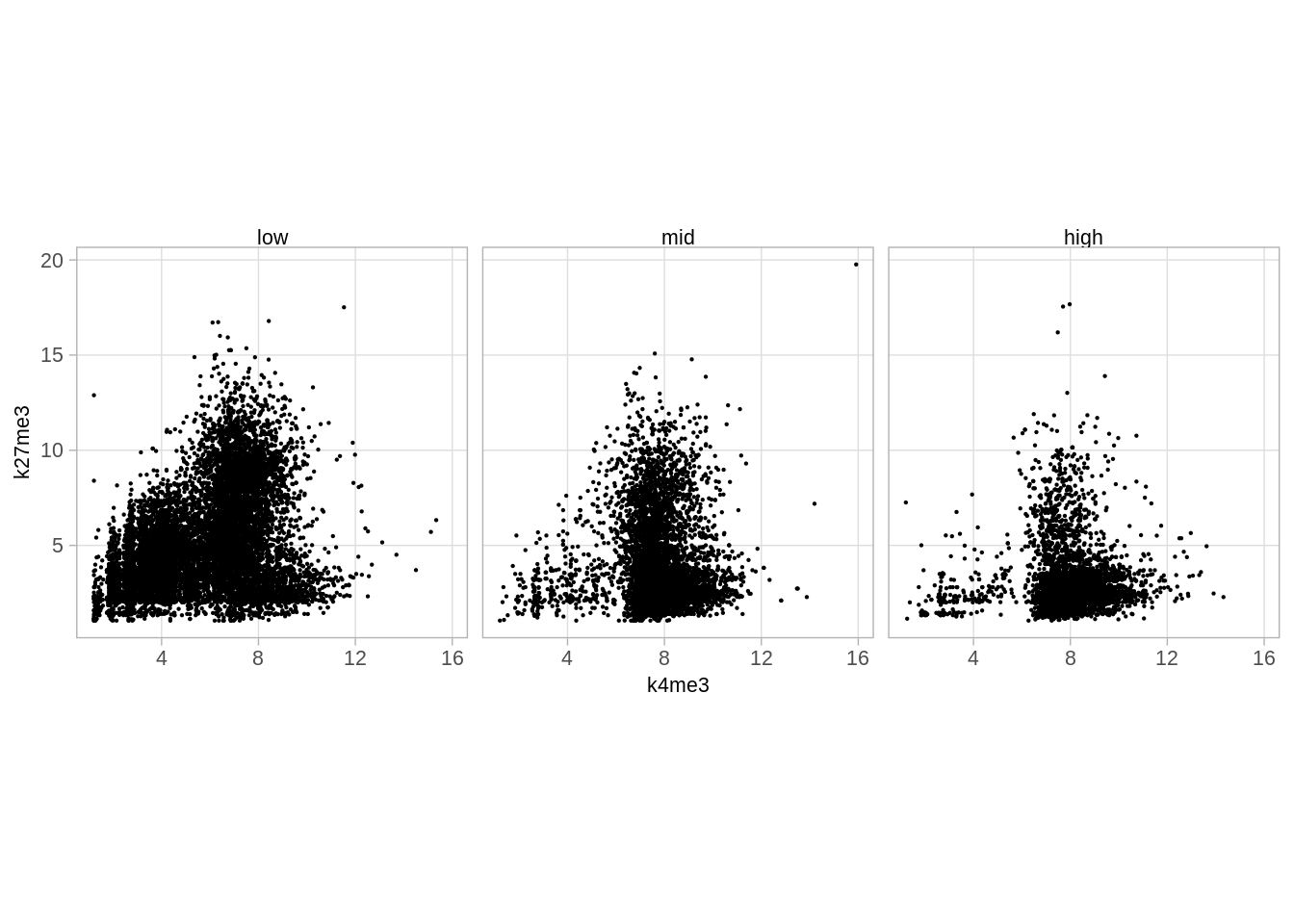
marks_expr %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("low", "mid", "high"))) %>%
filter(!is.na(k4me3)) %>%
count(expr)## # A tibble: 4 x 2
## expr n
## 1 low 11758
## 2 mid 6214
## 3 high 5040
## 4 <NA> 6### Figure S6D
options(repr.plot.width = 10, repr.plot.height = 4)
p_k4me3 <- marks_expr %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("low", "mid", "high"))) %>%
filter(!is.na(expr)) %>%
ggplot(aes(x = k4me3, color = expr, y = 1 - ..y..)) +
geom_vline(xintercept = 7, linetype = "dashed") +
stat_ecdf() +
ggsci::scale_color_npg(name = "Expression") +
ylab("% of promoters") +
scale_y_continuous(label = scales::percent)
p_k27me3 <- marks_expr %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("low", "mid", "high"))) %>%
filter(!is.na(expr)) %>%
ggplot(aes(x = k27me3, color = expr, y = 1 - ..y..)) +
geom_vline(xintercept = 7, linetype = "dashed") +
stat_ecdf() +
ggsci::scale_color_npg(name = "Expression") +
ylab("% of promoters") +
scale_y_continuous(label = scales::percent)
p_k4me3 + p_k27me3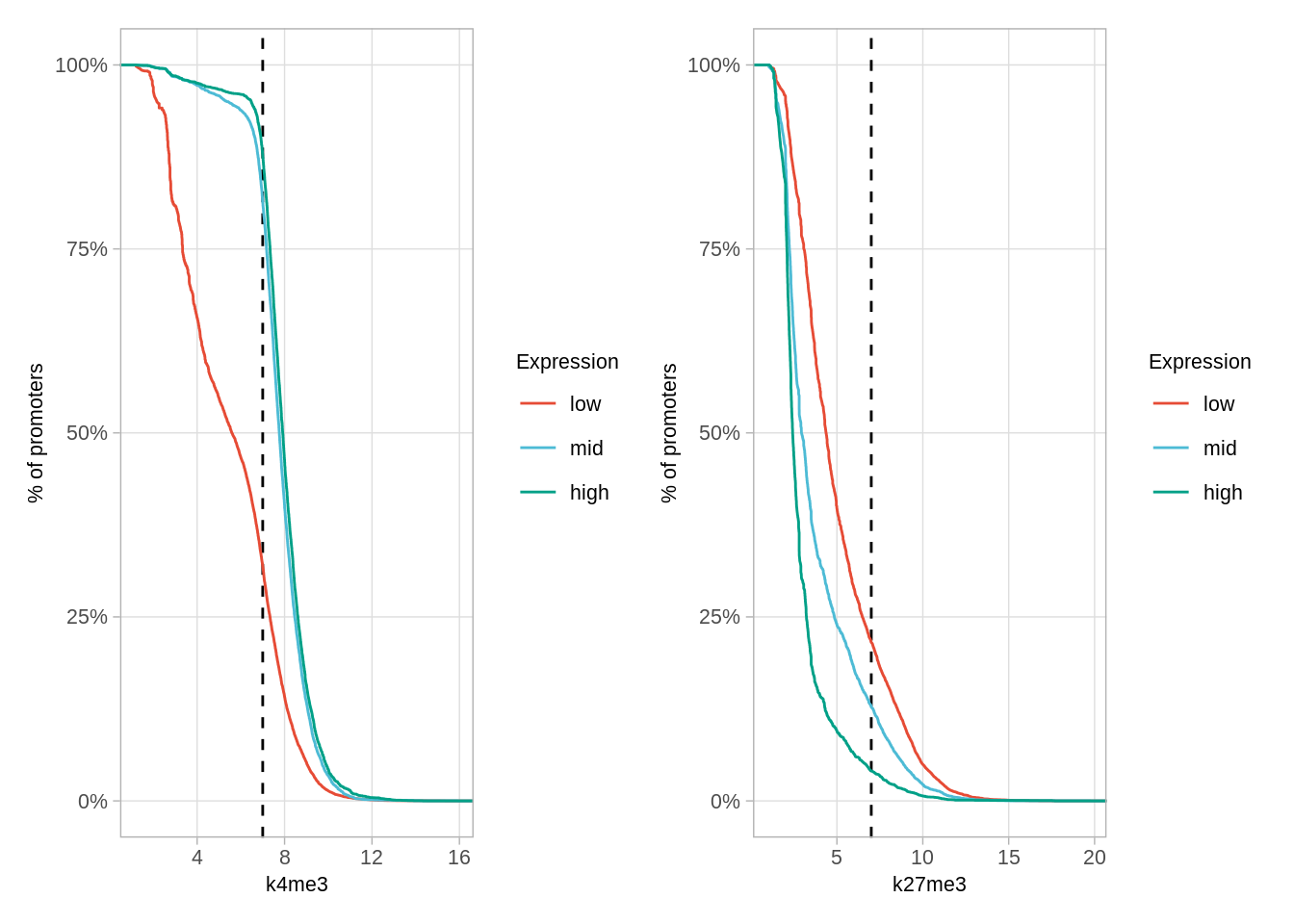
promoters_table %>%
filter(type == "none") %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("Low Expr.", "Mid Expr.", "High Expr."))) %>%
filter(!is.na(expr)) %>%
filter(ctrl.cov >= 30, tet_tko.cov >= 30) %>%
count(expr)## # A tibble: 3 x 2
## expr n
## 1 Low Expr. 3428
## 2 Mid Expr. 254
## 3 High Expr. 158### Figure S6E
options(repr.plot.width = 14, repr.plot.height = 4)
p_wt_tko_expr <- promoters_table %>%
filter(type == "none") %>%
mutate(expr = cut(wt_bulk, c(-17, -16, -14, 0), labels = c("Low Expr.", "Mid Expr.", "High Expr."))) %>%
filter(!is.na(expr)) %>%
filter(ctrl.cov >= 30, tet_tko.cov >= 30) %>%
ggplot(aes(x = ctrl, y = tet_tko)) +
geom_abline(linetype = "dashed") +
geom_point(size = 0.1) +
facet_wrap(~expr) +
theme(aspect.ratio = 1) +
vertical_labs() +
xlab("Ctrl.") +
ylab("TKO")
p_wt_tko_expr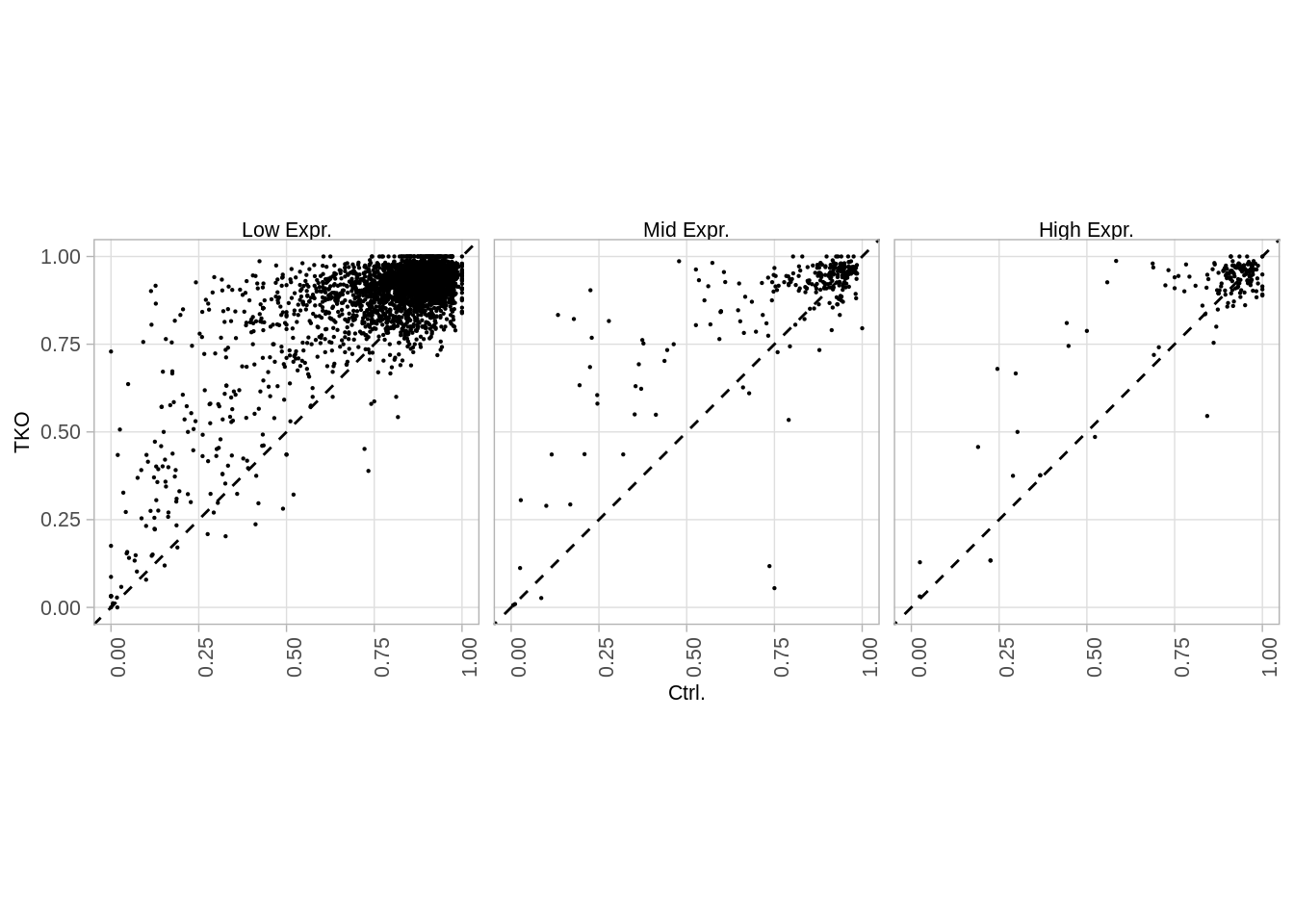
9.12 Putative enhancers
We extract CHIP data for methylation dips which do not have a known functional annotation (h4me3, k27me3 or promoters):
bg_intervs <- gintervals.diff(meth_dips, k4me3_peaks) %>%
gintervals.diff(k27me3_peaks) %>%
gintervals.diff(get_promoters())define_chip_vtracks()
meth_dips_chip <- bg_intervs %>%
gextract.left_join(
c(
"pmax(-log2(1-e10_limb_k4me1),-log2(1-e10_heart_k4me1),-log2(1-e10_fc_k4me1),-log2(1-e10_midbrain_k4me1),-log2(1-e10_hindbrain_k4me1),-log2(1-e10_forebrain_k4me1), na.rm=TRUE)",
"pmax(-log2(1-e10_limb_k27ac),-log2(1-e10_heart_k27ac),-log2(1-e10_midbrain_k27ac),-log2(1-e10_hindbrain_k27ac),-log2(1-e10_forebrain_k27ac),-log2(1-ecto_k27ac),-log2(1-endo_k27ac),-log2(1-meso_k27ac),-log2(1-ps_k27ac),-log2(1-e6_epi_k27ac),-log2(1-e6_ve_k27ac), na.rm=TRUE)"
),
iterator = ., intervals = ., colnames = c("k4me1", "k27ac")
) %>%
select(-(chrom1:end1)) %cache_df% here("output/meth_dips_chip.tsv") %>%
as_tibble()We extract the same data for all all other non-functionaly annotated regions:
chip_bg <- gextract(
c(
"pmax(-log2(1-e10_limb_k4me1),-log2(1-e10_heart_k4me1),-log2(1-e10_fc_k4me1),-log2(1-e10_midbrain_k4me1),-log2(1-e10_hindbrain_k4me1),-log2(1-e10_forebrain_k4me1), na.rm=TRUE)",
"pmax(-log2(1-e10_limb_k27ac),-log2(1-e10_heart_k27ac),-log2(1-e10_midbrain_k27ac),-log2(1-e10_hindbrain_k27ac),-log2(1-e10_forebrain_k27ac),-log2(1-ecto_k27ac),-log2(1-endo_k27ac),-log2(1-meso_k27ac),-log2(1-ps_k27ac),-log2(1-e6_epi_k27ac),-log2(1-e6_ve_k27ac), na.rm=TRUE)"
),
iterator = 50, intervals = gintervals.all() %>% gintervals.diff(meth_dips) %>% gintervals.diff(k4me3_peaks) %>% gintervals.diff(k27me3_peaks) %>% gintervals.diff(get_promoters()), colnames = c("k4me1", "k27ac")
) %>%
select(-intervalID) %>%
as_tibble() %cache_df% here("output/chip_bg.tsv") %>%
as_tibble()9.12.1 Figure S6F
We now compare the distribution of k4me1 and k27ac (enhancer markers) at methylation dips regions vs the background:
options(repr.plot.width = 14, repr.plot.height = 4)
p_k4me1 <- meth_dips_chip %>%
ggplot(aes(x = k4me1, y = 1 - ..y..)) +
stat_ecdf() +
stat_ecdf(data = chip_bg, color = "darkgray") +
ylab("% loci") +
scale_y_continuous(label = scales::percent)
p_k27ac <- meth_dips_chip %>%
ggplot(aes(x = k27ac, y = 1 - ..y..)) +
stat_ecdf() +
stat_ecdf(data = chip_bg, color = "darkgray") +
ylab("% loci") +
scale_y_continuous(label = scales::percent)
p_k4me1 + p_k27ac## Warning: Removed 2116563 rows containing non-finite values (stat_ecdf).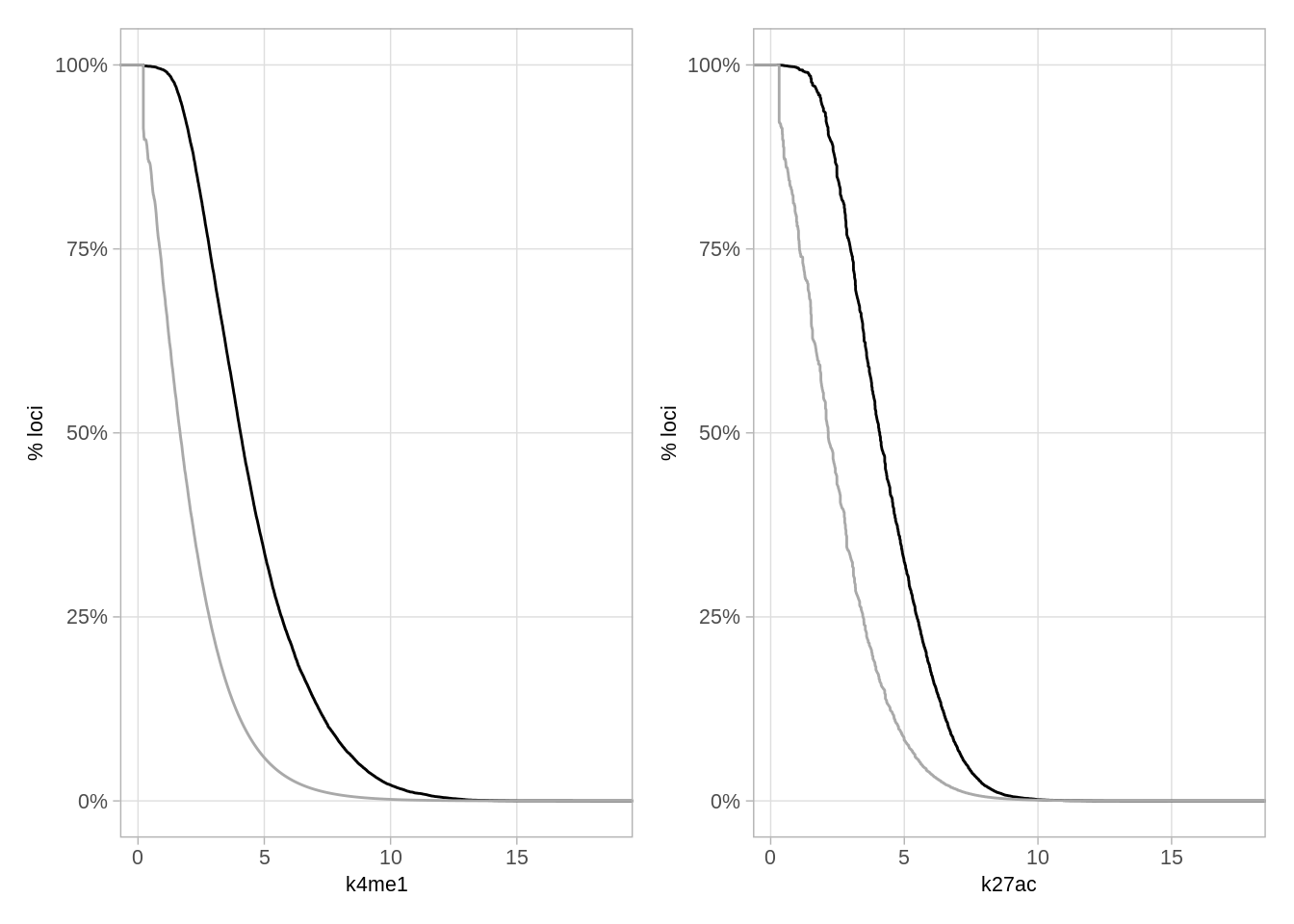
9.12.2 Enhancer methylation distribution (Figure 5H, Figure S6G)
d_bg_comp <- extract_meth_at_context(bg_intervs, iterator = "intervs.global.seq_CG", min_cov = 10) %cache_df% here("output/dips_bg_meth_comp.tsv") %>% as_tibble()## [1] 12720## [1] 34185options(repr.plot.width = 7, repr.plot.height = 7)
min_cov <- 10
plot_smooth_cpg_scatter(d_bg_comp, "ctrl", "tet_tko", min_cov, "Control", "Tet-TKO", colors = c("white", "lightgray", "lightblue", "blue", "red", "yellow"))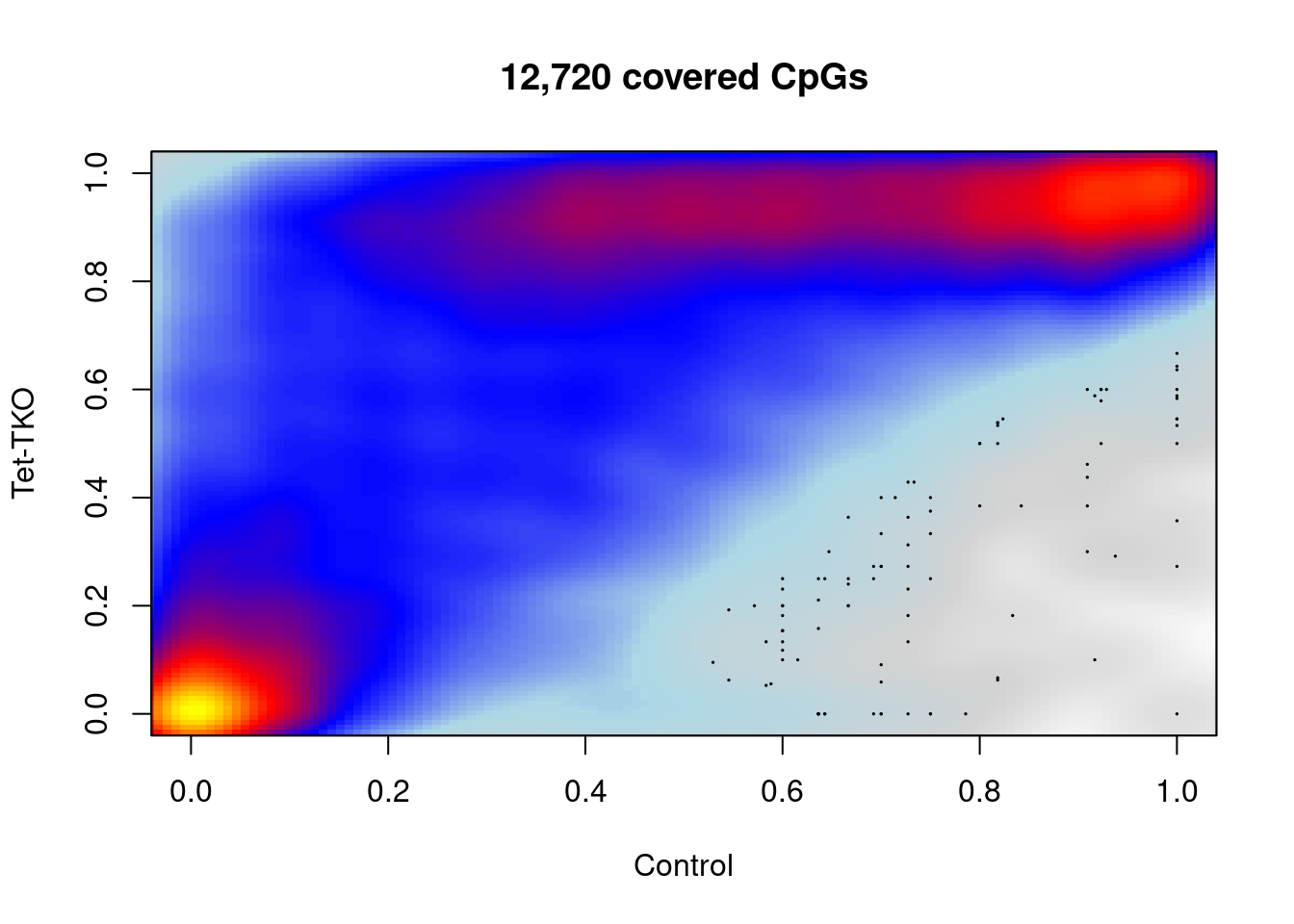
plot_smooth_cpg_scatter(d_bg_comp, "e7.5", "tet_tko", min_cov, "WT", "Tet-TKO", colors = c("white", "lightgray", "lightblue", "blue", "red", "yellow"))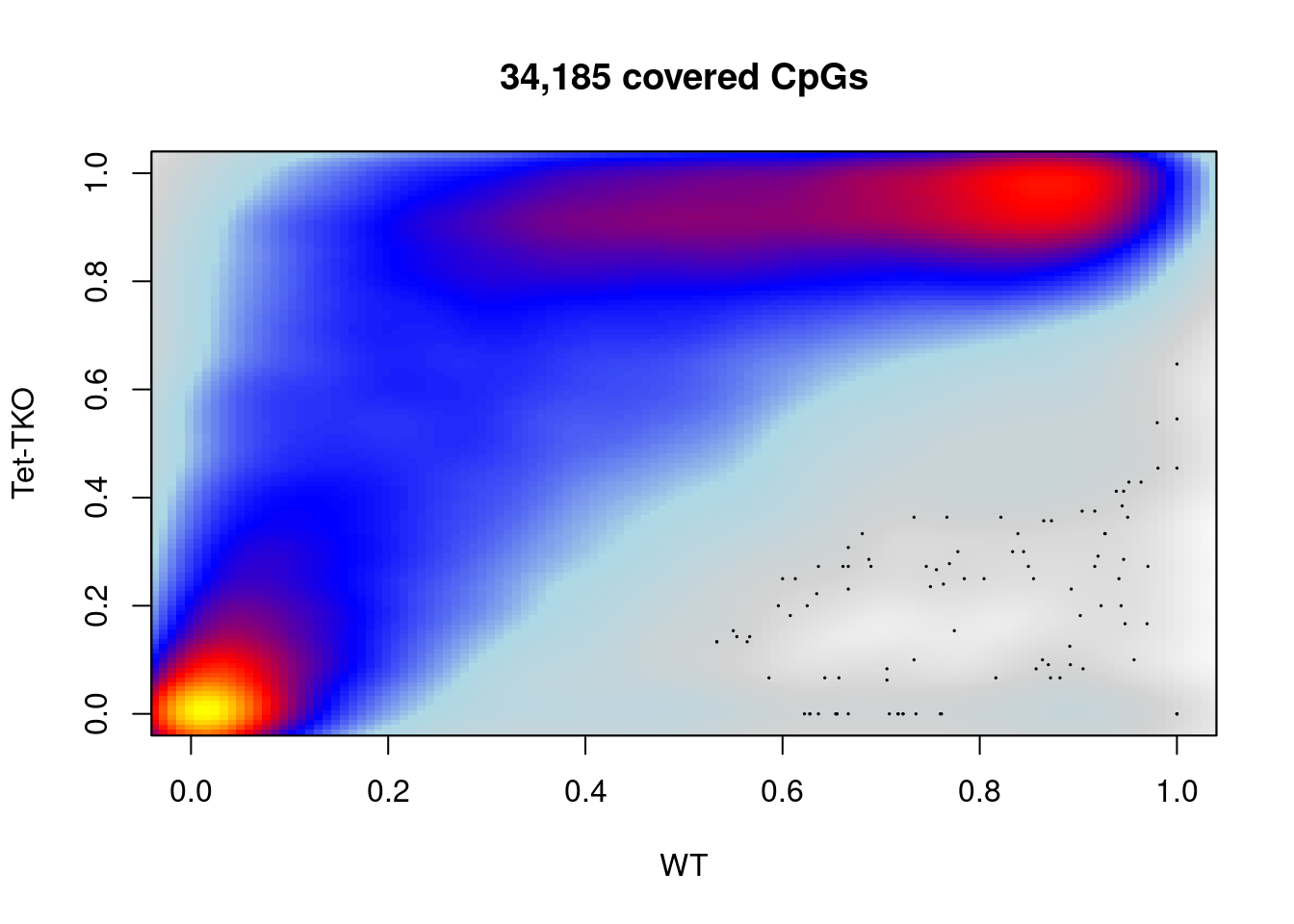
options(repr.plot.width = 5, repr.plot.height = 6)
plot_smooth_legend(c("white", "lightgray", "lightblue", "blue", "red", "yellow"))
9.12.3 Distribution of DNA methylation around the center of putative enhancers (Figure 5I)
df_dips_cpgs <- meth_df_comp_non_smoothed %>%
select(chrom:end, starts_with("ctrl"), starts_with("tet_tko")) %>%
gintervals.neighbors1(d_bg_comp %>% select(chrom, start, end, ctrl_dip = ctrl, wt_tet_tko = tet_tko) %>% mutate(strand = 1) %>% select(chrom, start, end, strand, everything()))options(repr.plot.width = 14, repr.plot.height = 7)
df <- df_dips_cpgs %>%
filter(abs(dist) <= 5e4) %>%
mutate(dist = cut(dist, breaks = seq(-500, 500, length.out = 6))) %>%
filter(!is.na(dist)) %>%
mutate(ctrl_dip = cut(ctrl_dip, c(0, 0.3, 0.7, 1), include.lowest = TRUE)) %>%
group_by(chrom1, start1, end1, ctrl_dip, dist) %>%
summarise_at(vars(ends_with(".cov"), ends_with(".meth")), sum, na.rm = TRUE) %>%
ungroup() %>%
mutate(Ctrl = ctrl.meth / ctrl.cov, TKO = tet_tko.meth / tet_tko.cov) %>%
filter(ctrl.cov >= 10, tet_tko.cov >= 10)
df %>% count(ctrl_dip)## # A tibble: 4 x 2
## ctrl_dip n
## 1 [0,0.3] 18815
## 2 (0.3,0.7] 16067
## 3 (0.7,1] 12567
## 4 <NA> 117473p_dips_boxp <- df %>%
pivot_longer(Ctrl:TKO, names_to = "type", values_to = "meth") %>%
ggplot(aes(x = dist, y = meth, fill = type)) +
geom_boxplot(lwd = 0.01, outlier.size = 0.001, fatten = 0.1) +
facet_grid(. ~ ctrl_dip) +
scale_fill_manual(name = "", values = c("Ctrl" = "#00468BFF", "TKO" = "#ED0000FF")) +
ylim(0, 1) +
xlab("") +
ylab("Methylation") +
vertical_labs()
p_dips_boxp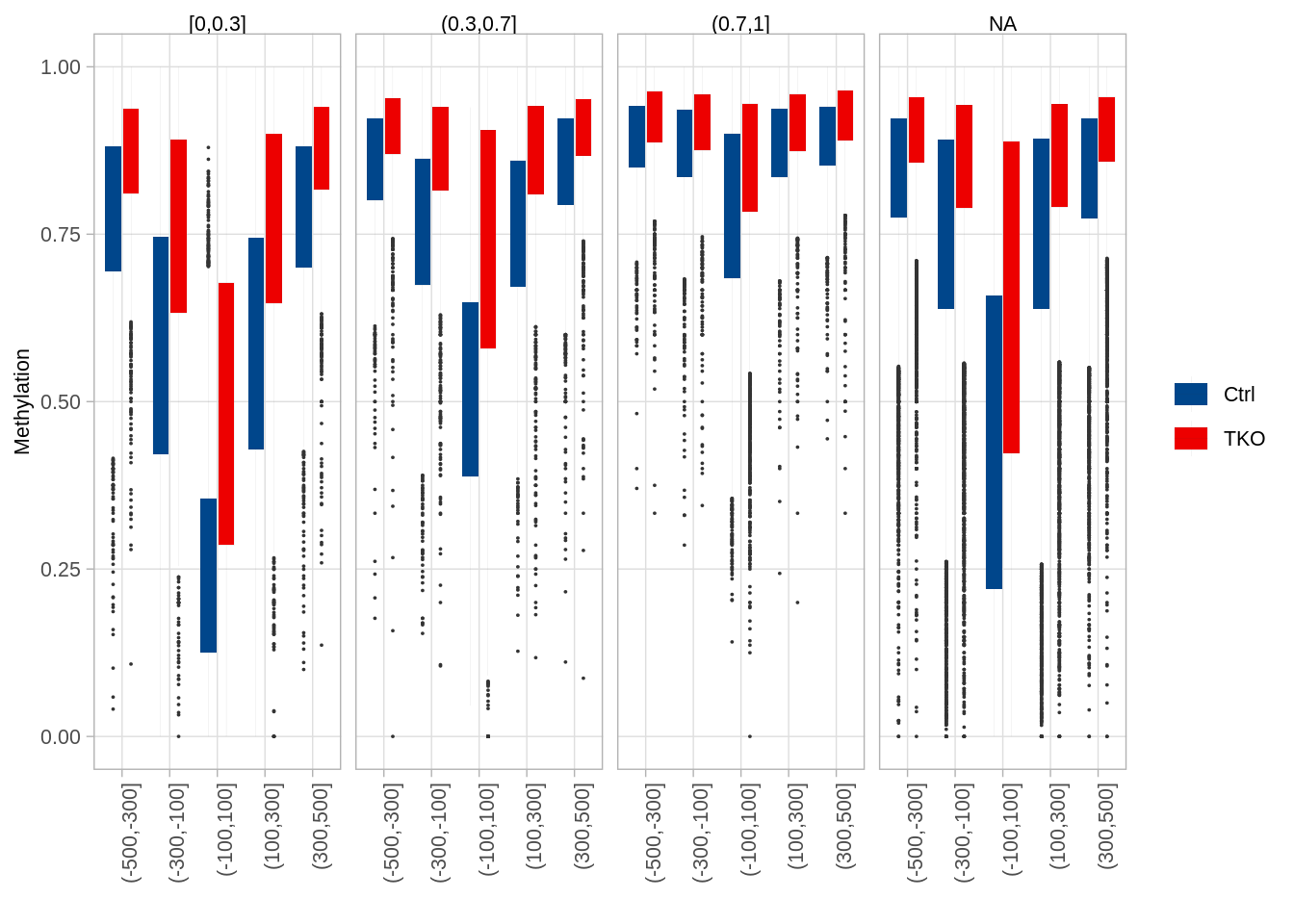
df %>%
filter(dist == "(-100,100]") %>%
group_by(ctrl_dip) %>%
summarise_at(vars(Ctrl:TKO), mean, na.rm = TRUE) %>%
mutate(diff = TKO - Ctrl) %>%
mutate_at(vars(Ctrl:TKO), scales::percent)## # A tibble: 4 x 4
## ctrl_dip Ctrl TKO diff
## 1 [0,0.3] 25.2% 48.7% 0.2350151
## 2 (0.3,0.7] 51.7% 72.5% 0.2077952
## 3 (0.7,1] 77.6% 84.2% 0.0659107
## 4 <NA> 44.4% 64.2% 0.1972279df %>%
filter(dist == "(-100,100]") %>%
group_by(ctrl_dip) %>%
summarise(p = ks.test(Ctrl, TKO)$p.value)## Warning in ks.test(Ctrl, TKO): p-value will be approximate in the presence of
## ties
## Warning in ks.test(Ctrl, TKO): p-value will be approximate in the presence of
## ties
## Warning in ks.test(Ctrl, TKO): p-value will be approximate in the presence of
## ties
## Warning in ks.test(Ctrl, TKO): p-value will be approximate in the presence of
## ties## # A tibble: 4 x 2
## ctrl_dip p
## 1 [0,0.3] 0
## 2 (0.3,0.7] 0
## 3 (0.7,1] 0
## 4 <NA> 09.13 Motifs (Figure S6H)
Due to the large size of the motif tracks, we provide here the script together with a pre-computed file:
dips_bg_expanded <- d_bg_comp %>%
distinct(chrom, start, end) %>%
mutate(start = start - 150, end = end + 150)
# motifs <- {
# library(gpwm)
# gpwm.extract_all("jolma_10bp", dips_bg_expanded, parallel=FALSE)
# }
motifs <- fread(here("data/dips_bg_expanded_motifs_jolma.tsv")) %>% as_tibble()motifs_df <- motifs %>%
gather("motif", "pwm", -(chrom:end)) %>%
filter(grepl("di_DBD", motif)) %>%
mutate(motif = gsub("jolma_10bp\\.", "", motif)) %>%
mutate(motif = gsub("_di_DBD", "", motif)) %>%
as_tibble()motifs_df <- gextract_meth(comb_tracks, names = comb_names, min_cov = min_cov, intervals = motifs_df, iterator = motifs_df, join_intervals = TRUE) %>%
select(-(chrom1:end1)) %>%
rename(control = ctrl, control.cov = ctrl.cov)Add global quantiles:
# global_quantiles <- gpwm.get_global_quantiles("jolma_10bp", size=10)
global_quantiles <- fread(here("data/motifs_global_quantiles.tsv")) %>% as_tibble()motifs_df_thresh <- global_quantiles %>%
filter(grepl("di_DBD", track)) %>%
mutate(motif = gsub("_di_DBD", "", track)) %>%
filter(quant == 0.995) %>%
select(motif, thresh = value) %>%
right_join(motifs_df) %>%
mutate(hit = pwm >= thresh)## Joining, by = "motif"motifs_summary <- motifs_df_thresh %>%
group_by(motif) %>%
mutate(diff = tet_tko - control) %>%
mutate(p.value = suppressWarnings(ks.test(diff[hit], diff[!hit])$p.value)) %>%
group_by(motif, hit) %>%
summarise(p.value = mean(p.value), n = n(), control = mean(control, na.rm = TRUE), tet_tko = mean(tet_tko, na.rm = TRUE)) %>%
ungroup() %>%
mutate(hit = ifelse(hit, "on", "off")) %>%
pivot_wider(names_from = "hit", values_from = c("n", "control", "tet_tko")) %>%
select(motif, n_off, control_off, tet_tko_off, n_on, control_on, tet_tko_on, `pval (of on diff vs off diff)` = p.value)9.13.1 Table S3
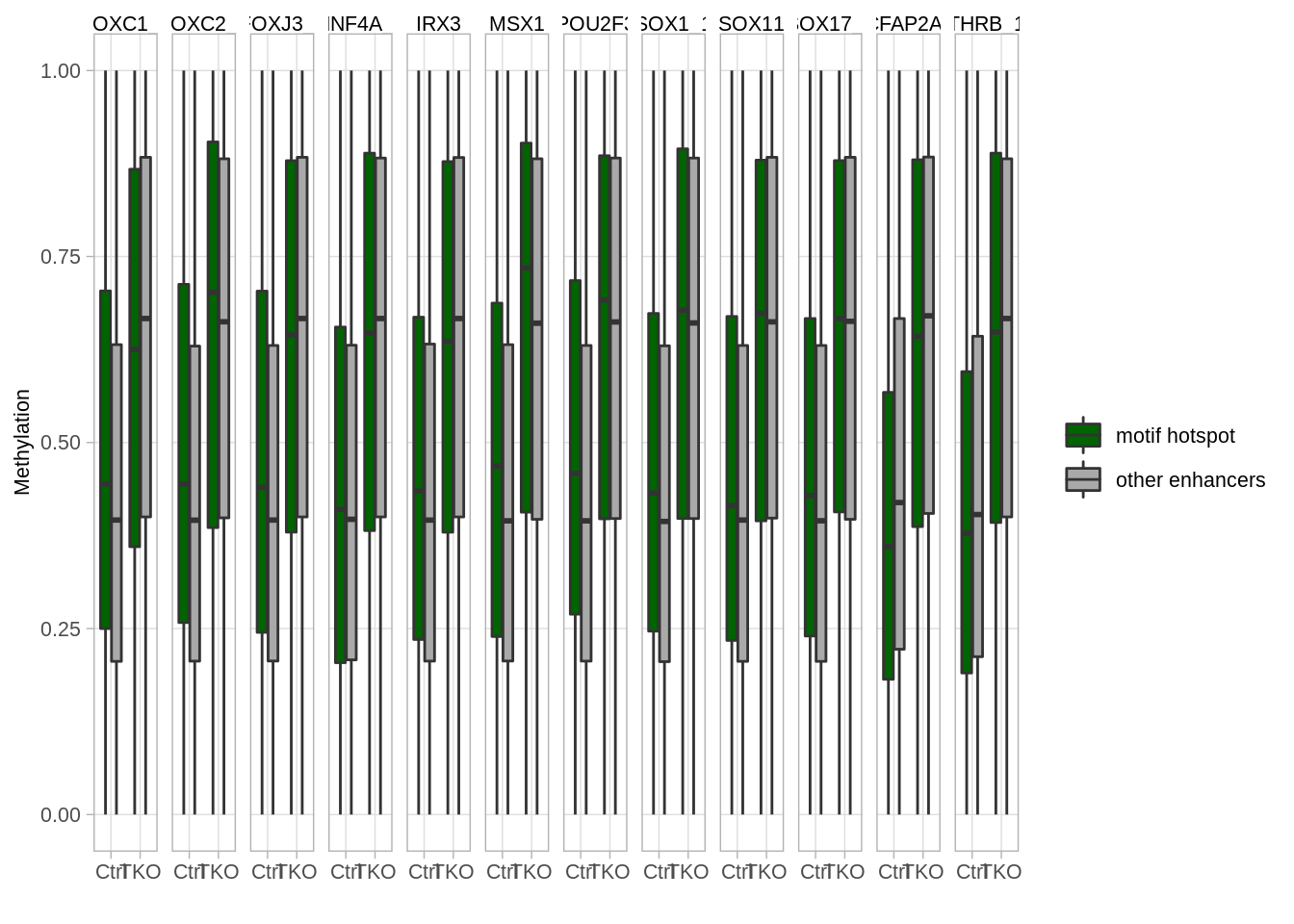
motifs_to_plot <- c("POU2F3", "FOXJ3_4", "SOX11", "SOX1", "TCFAP2A", "IRX3", "SOX17", "HNF4A", "THRB", "FOXC1", "FOXC2", "MSX1")
motifs_to_plot <- map_chr(motifs_to_plot, ~ motifs_df_thresh %>%
distinct(motif) %>%
filter(grepl(.x, motif)) %>%
slice(1) %>%
pull(motif))
motifs_to_plot## [1] "POU2F3" "FOXJ3_4" "SOX11" "SOX1_1" "TCFAP2A_1" "IRX3"
## [7] "SOX17_1" "HNF4A_1" "THRB_1" "FOXC1_1" "FOXC2_1" "MSX1"p_motifs_boxp <- motifs_df_thresh %>%
filter(motif %in% motifs_to_plot) %>%
select(-ends_with("cov")) %>%
rename(Ctrl = control, TKO = tet_tko) %>%
pivot_longer(names_to = "type", cols = c("Ctrl", "TKO"), values_to = "meth") %>%
mutate(hit = ifelse(hit, "motif hotspot", "other enhancers")) %>%
ggplot(aes(x = type, fill = hit, y = meth)) +
geom_boxplot() +
ylab("Methylation") +
xlab("") +
scale_fill_manual(name = "", values = rev(c("darkgray", "darkgreen"))) +
facet_grid(. ~ motif)
p_motifs_boxp