6 Epigenomic instability
source(here::here("scripts/init.R"))6.1 Compare features to each other
6.1 We change the direction (sign) of the
clockandMLin order for it to be aligned withMGscore, i.e. more progressed always equals higher score. This is implemented also inget_all_features()function.
feats_df_fixed <- fread(here("data/epigenomic_features.tsv")) %>% mutate(ML = -ML, clock = -clock) %>% as_tibble()6.1.0.1 Figure 2E
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER+"], feats_df_fixed$ML[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = -0.166831861226654p_MG_ML_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=MG, y=ML, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("ML epigenomic instability") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER-"], feats_df_fixed$ML[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = -0.382303128930548p_MG_ML_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=MG, y=ML, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("ML epigenomic instability") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_MG_ML_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_MG_ML_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))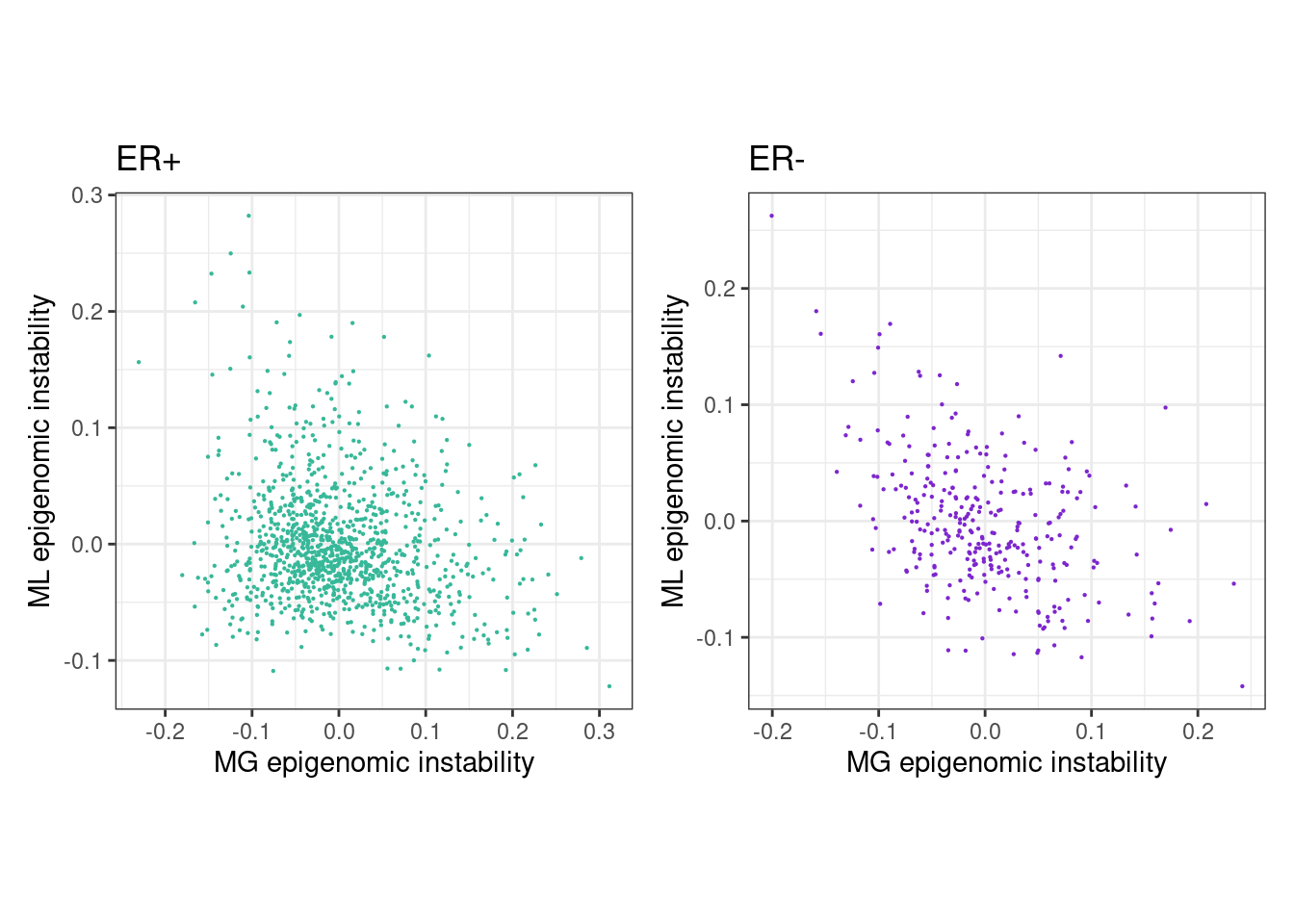
6.1.0.2 Extended Data Figure 6A
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER+"], feats_df_fixed$clock[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = -0.162654292782067p_MG_clock_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=MG, y=clock, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("Clock") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER-"], feats_df_fixed$clock[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = -0.28563668716636p_MG_clock_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=MG, y=clock, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("Clock") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_MG_clock_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_MG_clock_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))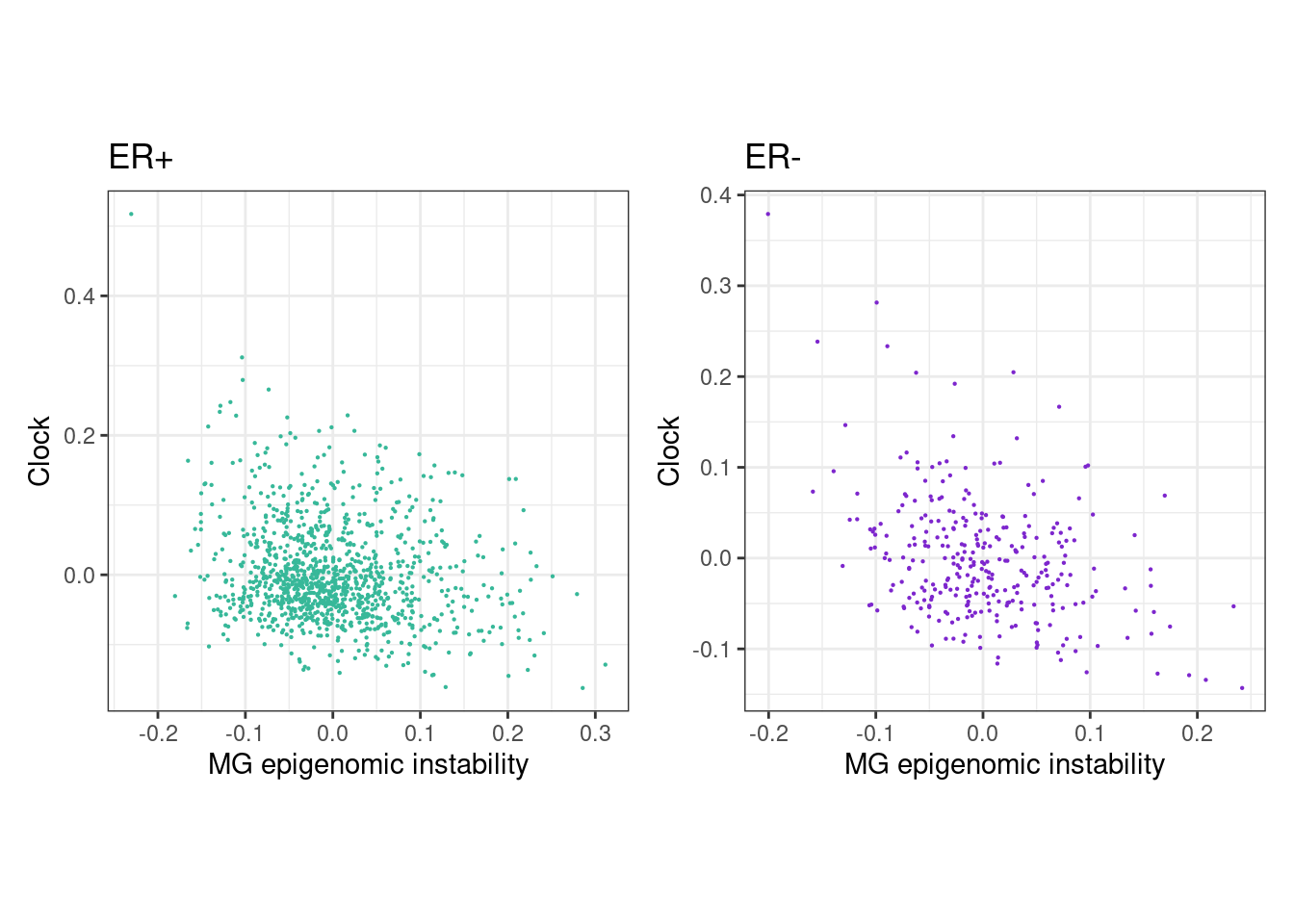
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER+"], feats_df_fixed$clock[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = 0.353548126048103p_ML_clock_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=ML, y=clock, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("Clock") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER-"], feats_df_fixed$clock[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = 0.64775559075671p_ML_clock_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=ML, y=clock, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("Clock") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_ML_clock_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_ML_clock_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))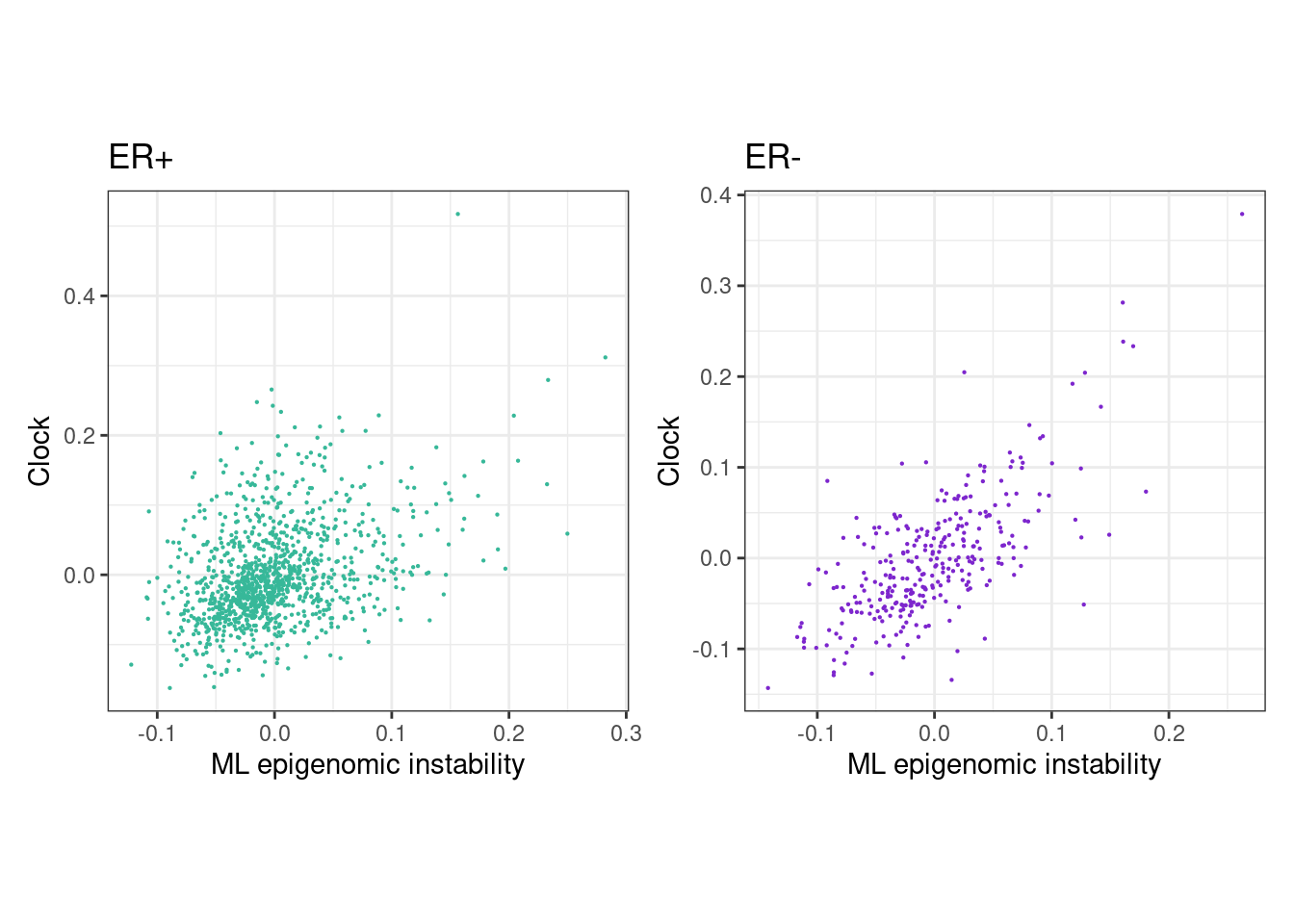
6.1.0.3 Extended Data Figure 6B
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER+"], feats_df_fixed$immune[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = 0.0256502058498626p_MG_immune_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=MG, y=immune, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("immune") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER-"], feats_df_fixed$immune[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = -0.0505710341049502p_MG_immune_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=MG, y=immune, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("immune") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_MG_immune_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_MG_immune_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))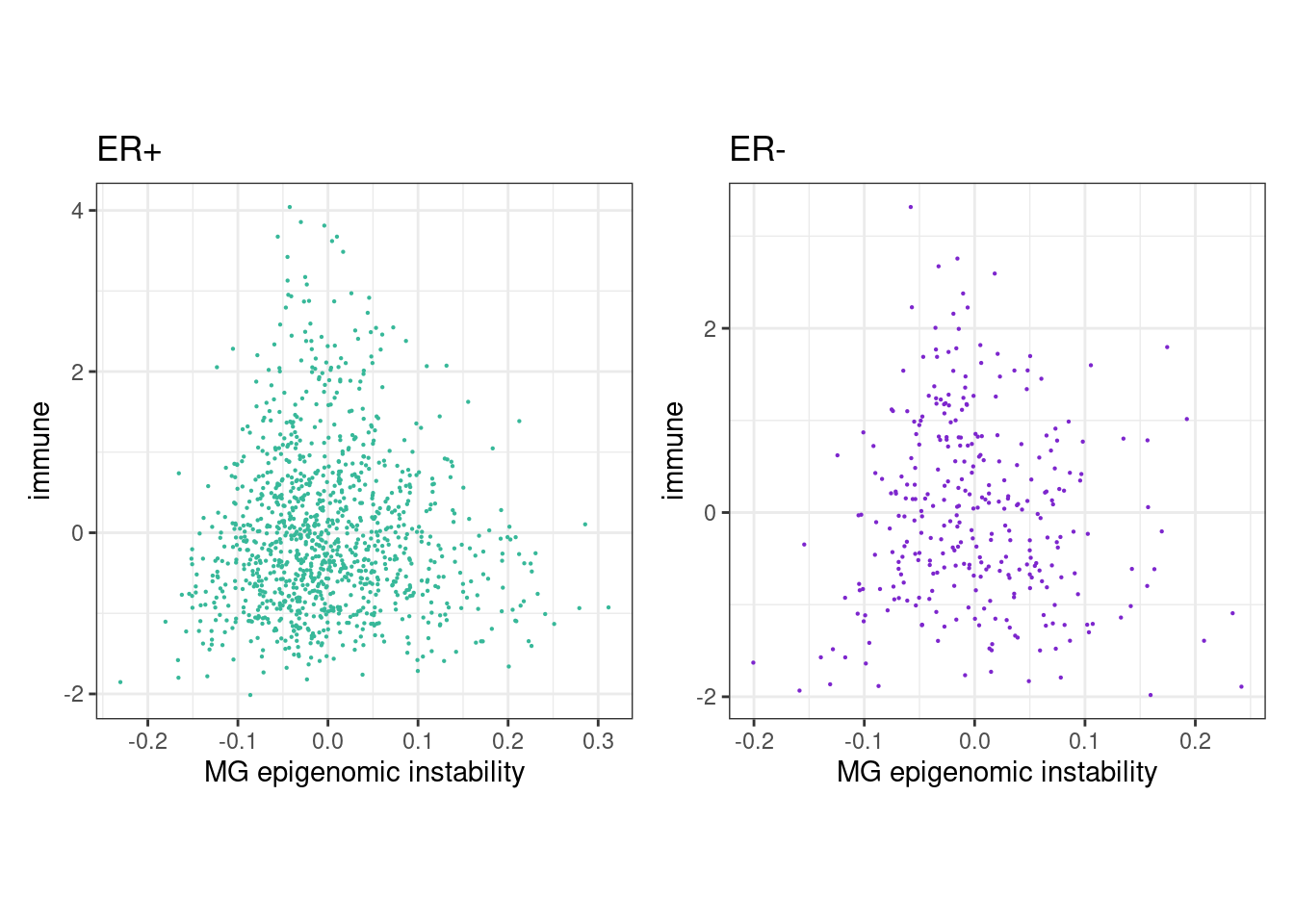
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER+"], feats_df_fixed$immune[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = 0.0182424884995677p_ML_immune_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=ML, y=immune, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("immune") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER-"], feats_df_fixed$immune[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = -0.0483692724136299p_ML_immune_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=ML, y=immune, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("immune") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_ML_immune_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_ML_immune_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))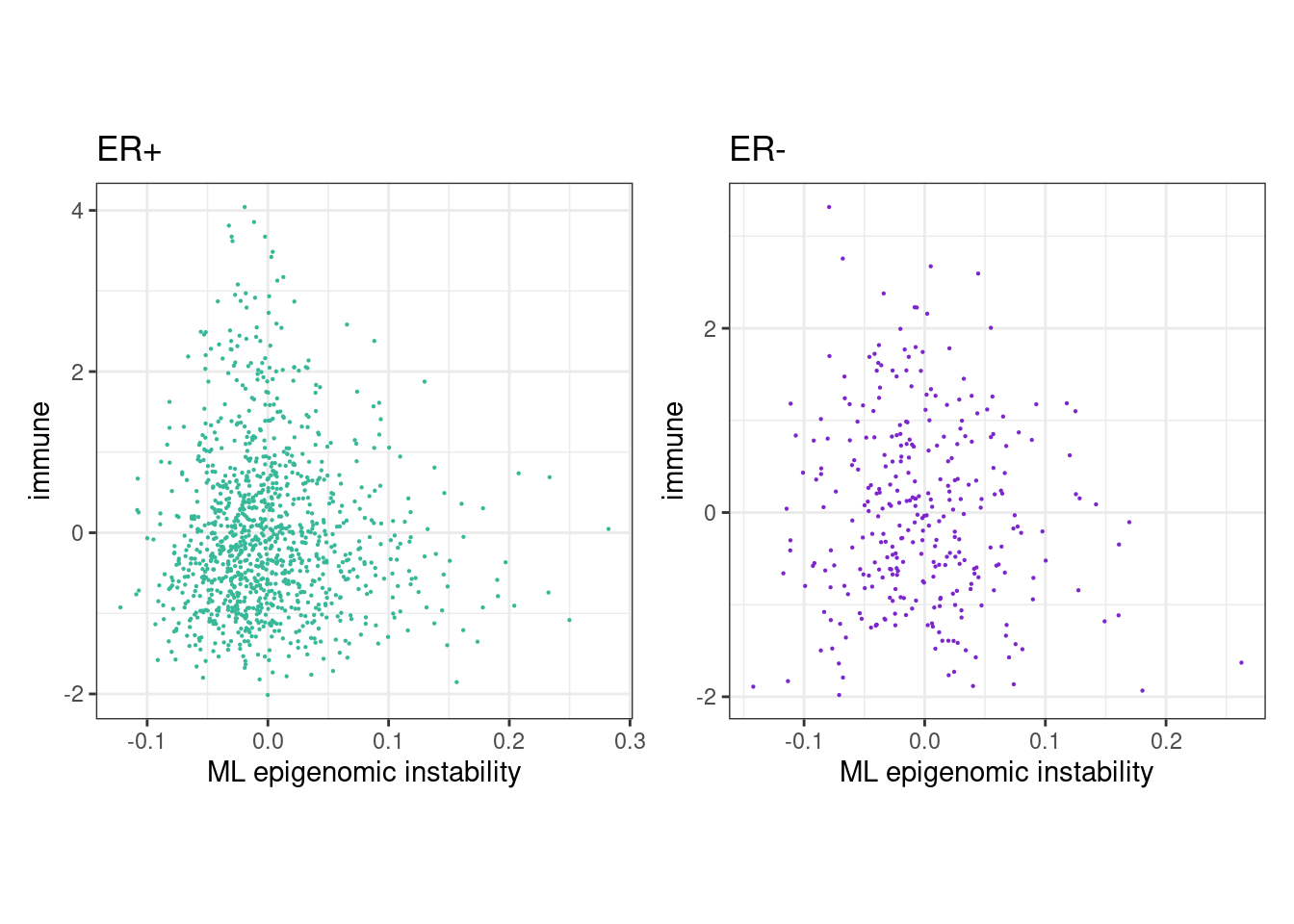
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER+"], feats_df_fixed$caf[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = 0.0201120969323973p_MG_caf_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=MG, y=caf, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("caf") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$MG[feats_df_fixed$ER == "ER-"], feats_df_fixed$caf[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = -0.00276791172007093p_MG_caf_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=MG, y=caf, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("MG epigenomic instability") +
ylab("caf") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_MG_caf_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_MG_caf_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))
options(repr.plot.width = 8, repr.plot.height = 4)
rho_pos <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER+"], feats_df_fixed$caf[feats_df_fixed$ER == "ER+"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER+ = {rho_pos}"))## rho ER+ = 0.0336682666159654p_ML_caf_scatter_ER_positive <- feats_df_fixed %>%
filter(ER == "ER+") %>%
ggplot(aes(x=ML, y=caf, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("caf") +
guides(color=FALSE) +
ggtitle("ER+")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.rho_neg <- cor(feats_df_fixed$ML[feats_df_fixed$ER == "ER-"], feats_df_fixed$caf[feats_df_fixed$ER == "ER-"], use="pairwise.complete.obs", method="spearman")
message(glue("rho ER- = {rho_neg}"))## rho ER- = 0.0020186843607852p_ML_caf_scatter_ER_negative <- feats_df_fixed %>%
filter(ER == "ER-") %>%
ggplot(aes(x=ML, y=caf, color=ER)) +
geom_point(size=0.1) +
scale_color_manual(values = annot_colors$ER1) +
theme(aspect.ratio = 1) +
xlab("ML epigenomic instability") +
ylab("caf") +
guides(color=FALSE) +
ggtitle("ER-")## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.(p_ML_caf_scatter_ER_positive + theme_bw() + theme(aspect.ratio = 1)) + (p_ML_caf_scatter_ER_negative + theme_bw() + theme(aspect.ratio = 1))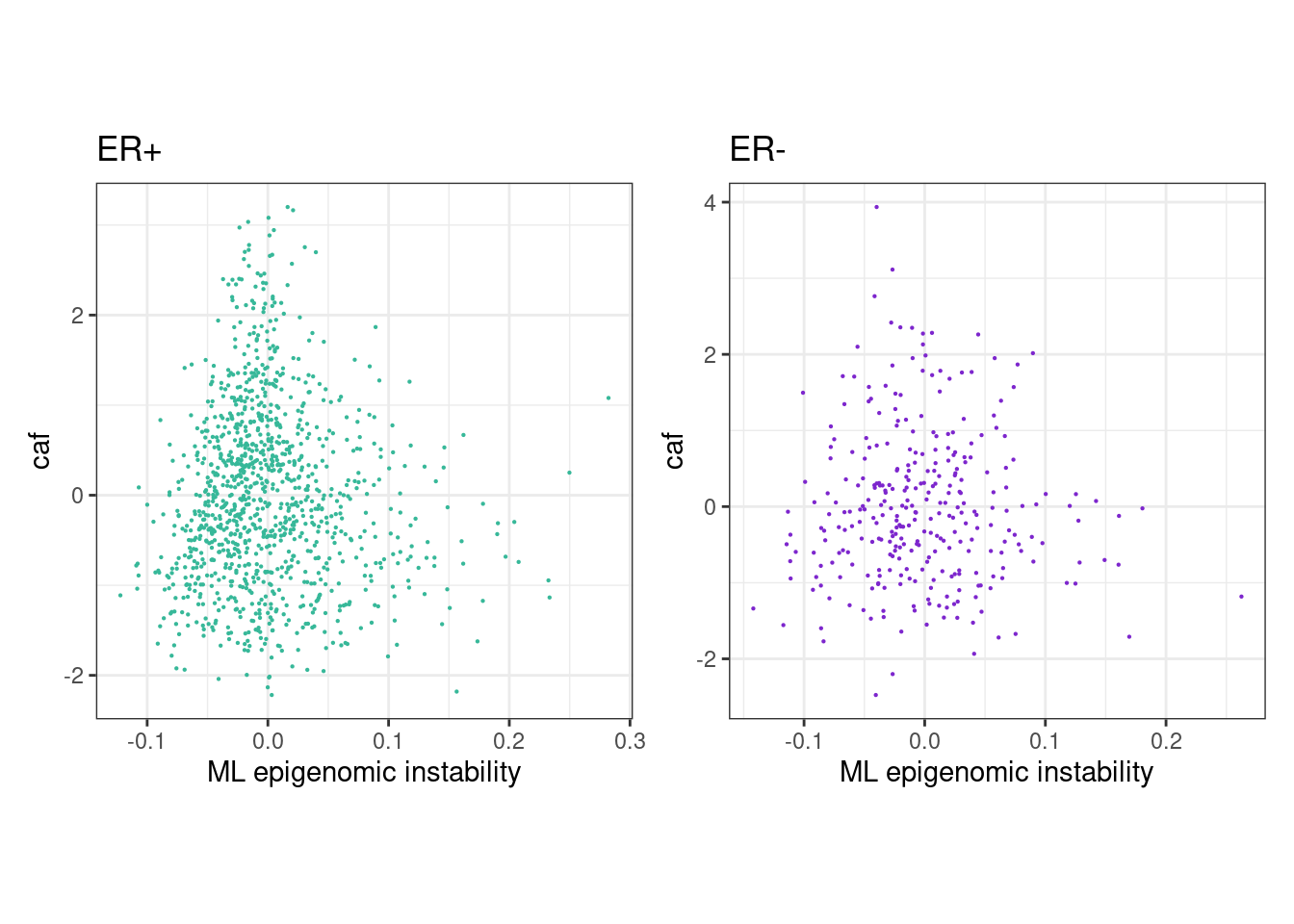
6.2 Annotate epignomic instability scores
feats <- get_all_features()
nbins <- 5
df <- feats %>%
mutate(
clock = cut(clock, quantile(clock, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
immune = cut(immune, quantile(immune, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
caf = cut(caf, quantile(caf, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
immune.meth = cut(immune.meth, quantile(immune.meth, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
caf.meth = cut(caf.meth, quantile(caf.meth, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
MG = cut(MG, quantile(MG, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins),
ML = cut(ML, quantile(ML, 0:nbins/nbins, na.rm=TRUE), include.lowest=TRUE, labels=1:nbins)) %>%
left_join(samp_data %>% select(samp, stage, grade), by = "samp") %>%
mutate(stage = ifelse(stage %in% c(0, "DCIS", 1), "0-1", stage)) %>%
mutate(stage = ifelse(ER == "normal", "N", stage)) %>%
mutate(grade = ifelse(ER == "normal", "N", grade))df_pval <- df %>% filter(ER %in% c("ER+", "ER-")) %>% gather("feat", "bin", -samp, -ER, -stage, -grade) %>% group_by(ER, feat) %>% summarise(grade_pval = chisq.test(bin, grade)$p.value, stage_pval = chisq.test(bin, stage)$p.value) %>% mutate(signif_grade = case_when(grade_pval < 0.0001 ~ "****", grade_pval < 0.001 ~ "***", grade_pval < 0.01 ~ "**", grade_pval < 0.05 ~ "*"), signif_stage = case_when(stage_pval < 0.0001 ~ "****", stage_pval < 0.001 ~ "***", stage_pval < 0.01 ~ "**", stage_pval < 0.05 ~ "*"))## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, grade): Chi-squared approximation may be incorrect## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrect
## Warning in chisq.test(bin, stage): Chi-squared approximation may be incorrectas.data.frame(df_pval)## ER feat grade_pval stage_pval signif_grade signif_stage
## 1 ER- caf 2.264293e-04 0.001946125 *** **
## 2 ER- caf.meth 3.081915e-03 0.209690818 ** <NA>
## 3 ER- clock 6.348596e-01 0.070940655 <NA> <NA>
## 4 ER- immune 3.712915e-01 0.580276177 <NA> <NA>
## 5 ER- immune.meth 3.051790e-01 0.102242249 <NA> <NA>
## 6 ER- MG 5.763868e-01 0.042991948 <NA> *
## 7 ER- ML 6.479486e-01 0.873719290 <NA> <NA>
## 8 ER+ caf 6.512545e-05 0.060334718 **** <NA>
## 9 ER+ caf.meth 1.267372e-05 0.070515621 **** <NA>
## 10 ER+ clock 1.471181e-01 0.572271776 <NA> <NA>
## 11 ER+ immune 6.363196e-04 0.457202009 *** <NA>
## 12 ER+ immune.meth 5.579626e-03 0.293001516 ** <NA>
## 13 ER+ MG 1.588818e-06 0.099708353 **** <NA>
## 14 ER+ ML 5.428637e-10 0.230225194 **** <NA>df_pval %>% filter(grade_pval <= 0.05)## # A tibble: 8 x 6
## # groups: ER
## ER feat grade_pval stage_pval signif_grade signif_stage
## 1 ER- caf 0.00022642925 0.001946125 *** **
## 2 ER- caf.meth 0.00308191469 0.209690818 ** <NA>
## 3 ER+ caf 0.00006512545 0.060334718 **** <NA>
## 4 ER+ caf.meth 0.00001267372 0.070515621 **** <NA>
## 5 ER+ immune 0.00063631960 0.457202009 *** <NA>
## 6 ER+ immune.meth 0.00557962596 0.293001516 ** <NA>
## # ... with 2 more rowsdf_pval %>% filter(stage_pval <= 0.05)## # A tibble: 2 x 6
## # groups: ER
## ER feat grade_pval stage_pval signif_grade signif_stage
## 1 ER- caf 0.0002264293 0.001946125 *** **
## 2 ER- MG 0.5763867809 0.042991948 <NA> *6.2.0.1 Extended Data Figure 6D
options(repr.plot.width = 6, repr.plot.height = 4)
p_stage_MG <- df %>%
filter(ER == "ER+") %>%
mutate(stage = factor(stage, levels = c("N", "0-1", "2", "3", "4"))) %>%
filter(!is.na(stage)) %>%
count(stage, MG) %>%
group_by(MG) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=MG, y=p, fill=stage)) +
geom_col() +
scale_fill_manual(name = "Stage", values = c("N" = "gray", "0-1" = "black", "2" = "blue", "3" = "red", "4" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_stage_ML <- df %>%
filter(ER == "ER+") %>%
mutate(stage = factor(stage, levels = c("N", "0-1", "2", "3", "4"))) %>%
filter(!is.na(stage)) %>%
count(stage, ML) %>%
group_by(ML) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=ML, y=p, fill=stage)) +
geom_col() +
scale_fill_manual(name = "Stage", values = c("N" = "gray", "0-1" = "black", "2" = "blue", "3" = "red", "4" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_stage_MG + theme_bw()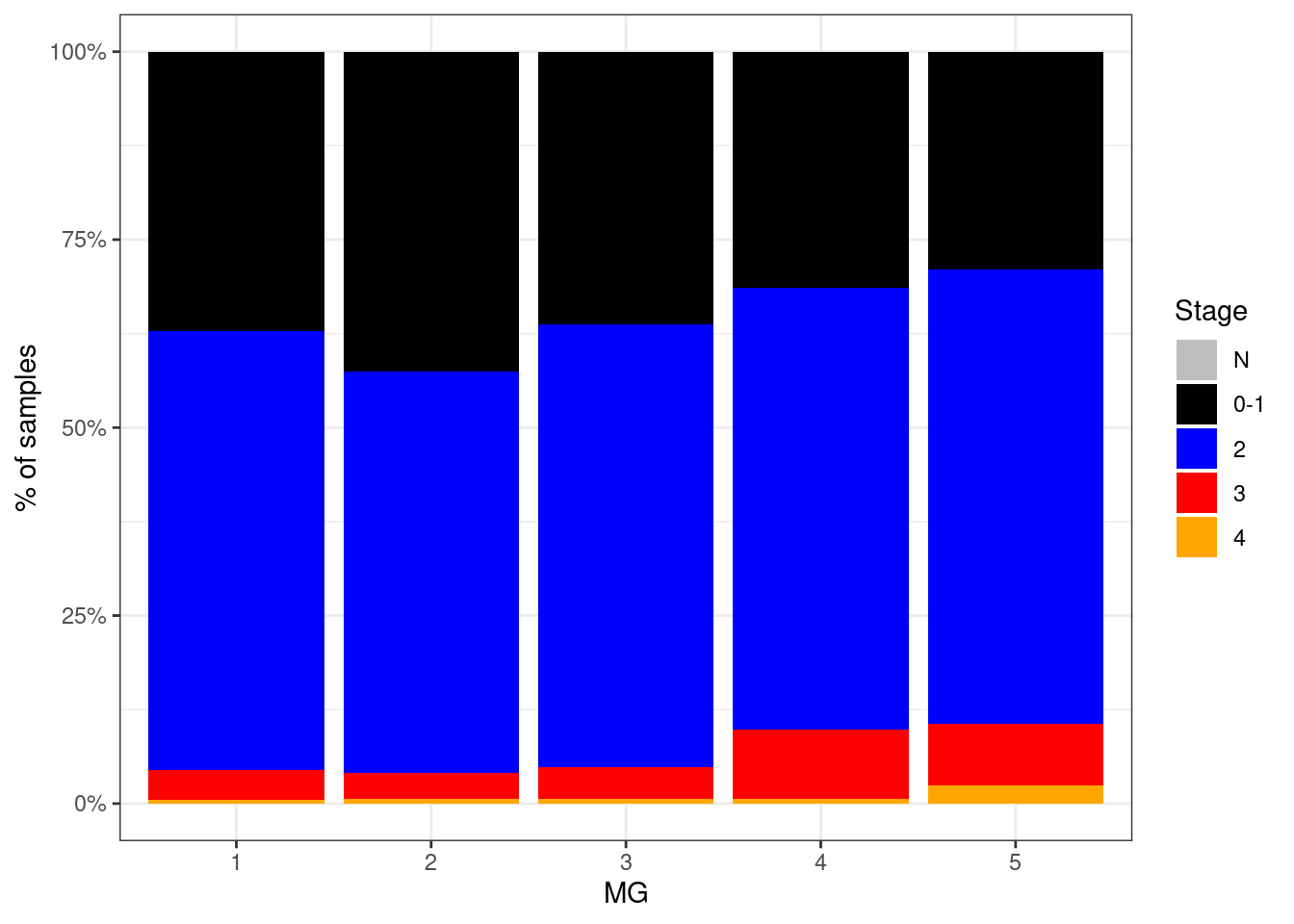
p_stage_ML + theme_bw()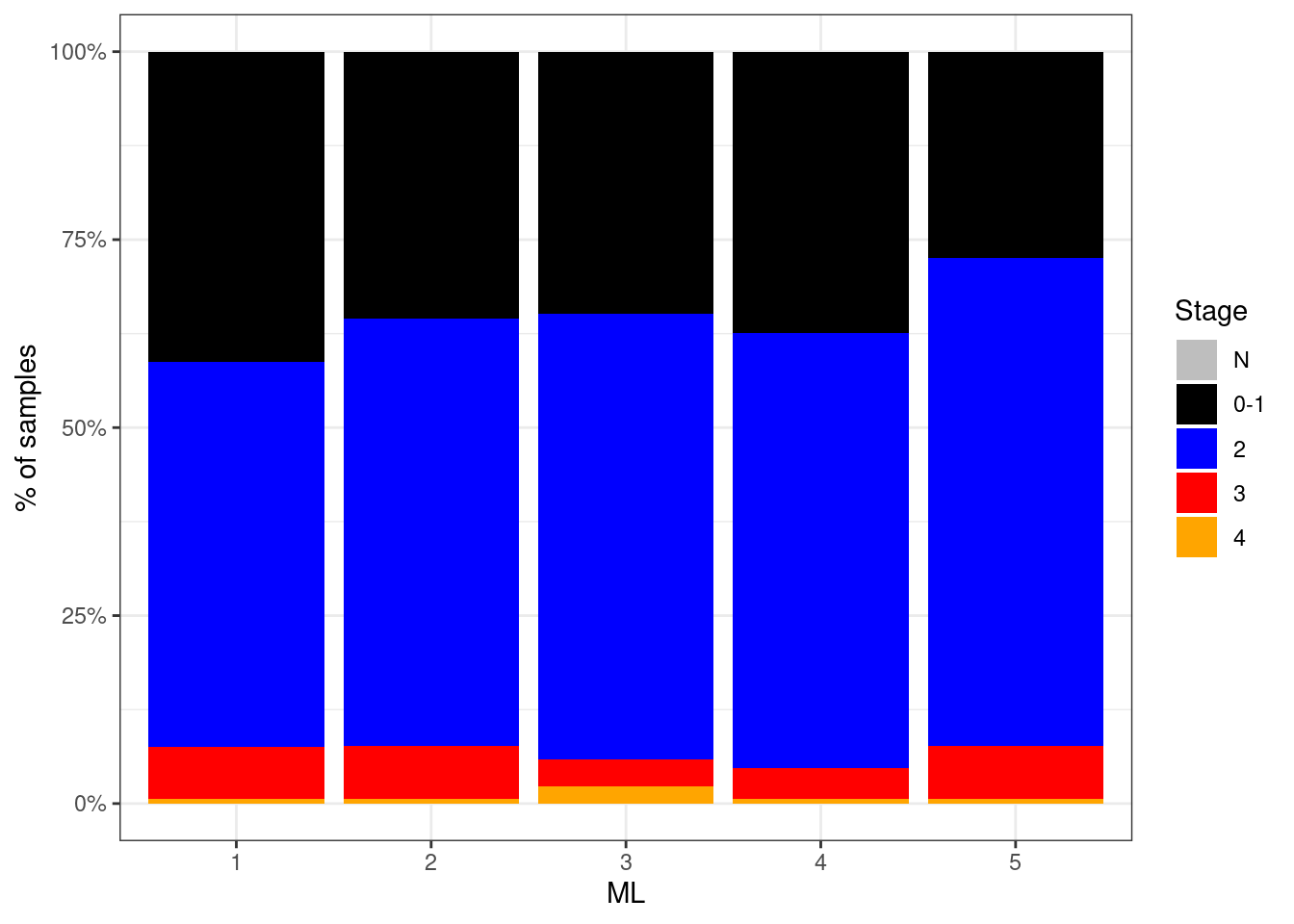
Same for ER-:
options(repr.plot.width = 6, repr.plot.height = 4)
p_grade_MG_neg <- df %>%
filter(ER == "ER-") %>%
mutate(grade = factor(grade, levels = c("N", "1", "2", "3"))) %>%
filter(!is.na(grade)) %>%
count(grade, MG) %>%
group_by(MG) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=MG, y=p, fill=grade)) +
geom_col() +
scale_fill_manual(name = "Grade", values = c("N" = "gray", "1" = "darkblue", "2" = "red", "3" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_grade_ML_neg <- df %>%
filter(ER == "ER-") %>%
mutate(grade = factor(grade, levels = c("N", "1", "2", "3"))) %>%
filter(!is.na(grade)) %>%
count(grade, ML) %>%
group_by(ML) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=ML, y=p, fill=grade)) +
geom_col() +
scale_fill_manual(name = "Grade", values = c("N" = "gray", "1" = "darkblue", "2" = "red", "3" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_grade_MG_neg + theme_bw()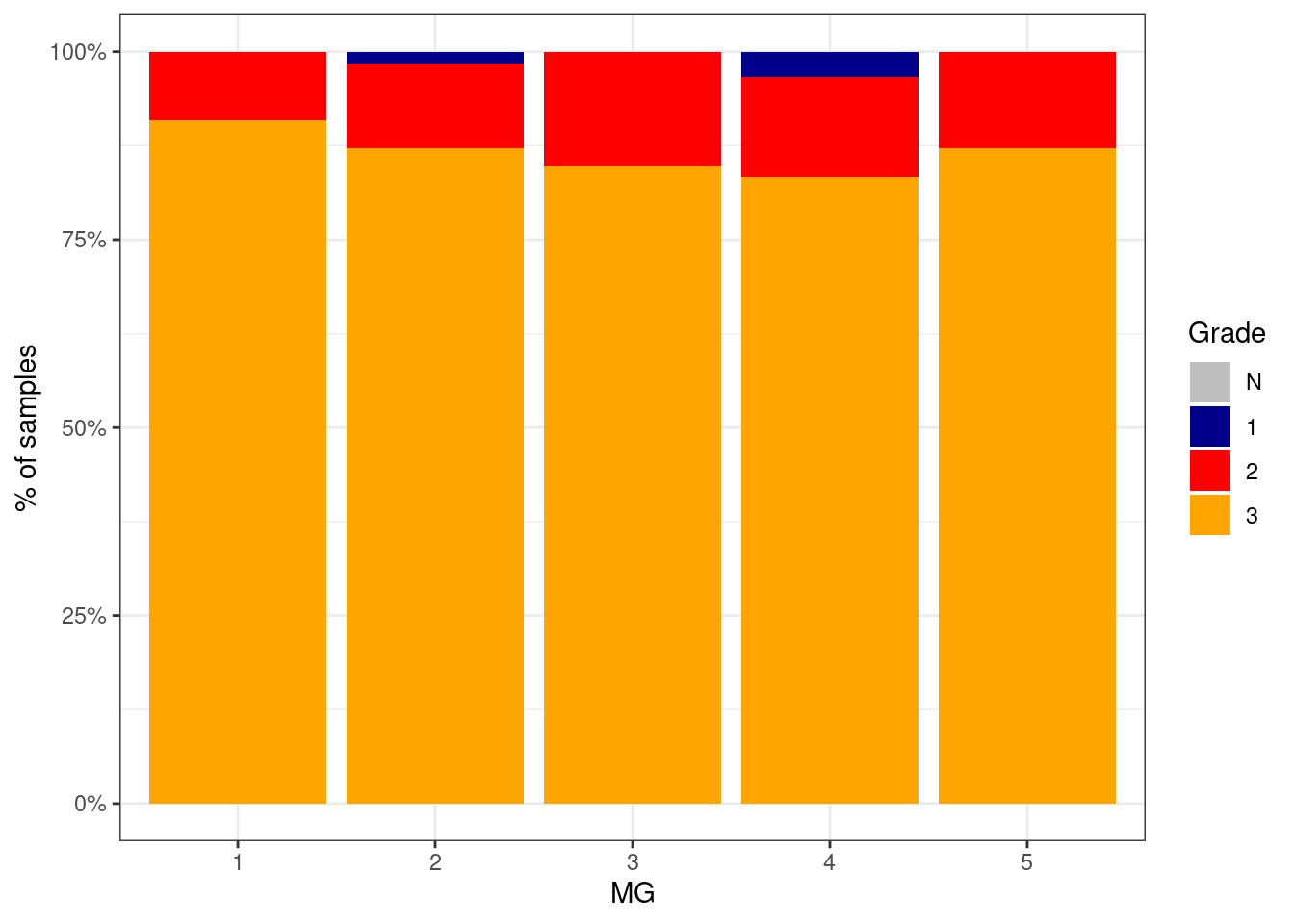
p_grade_ML_neg + theme_bw()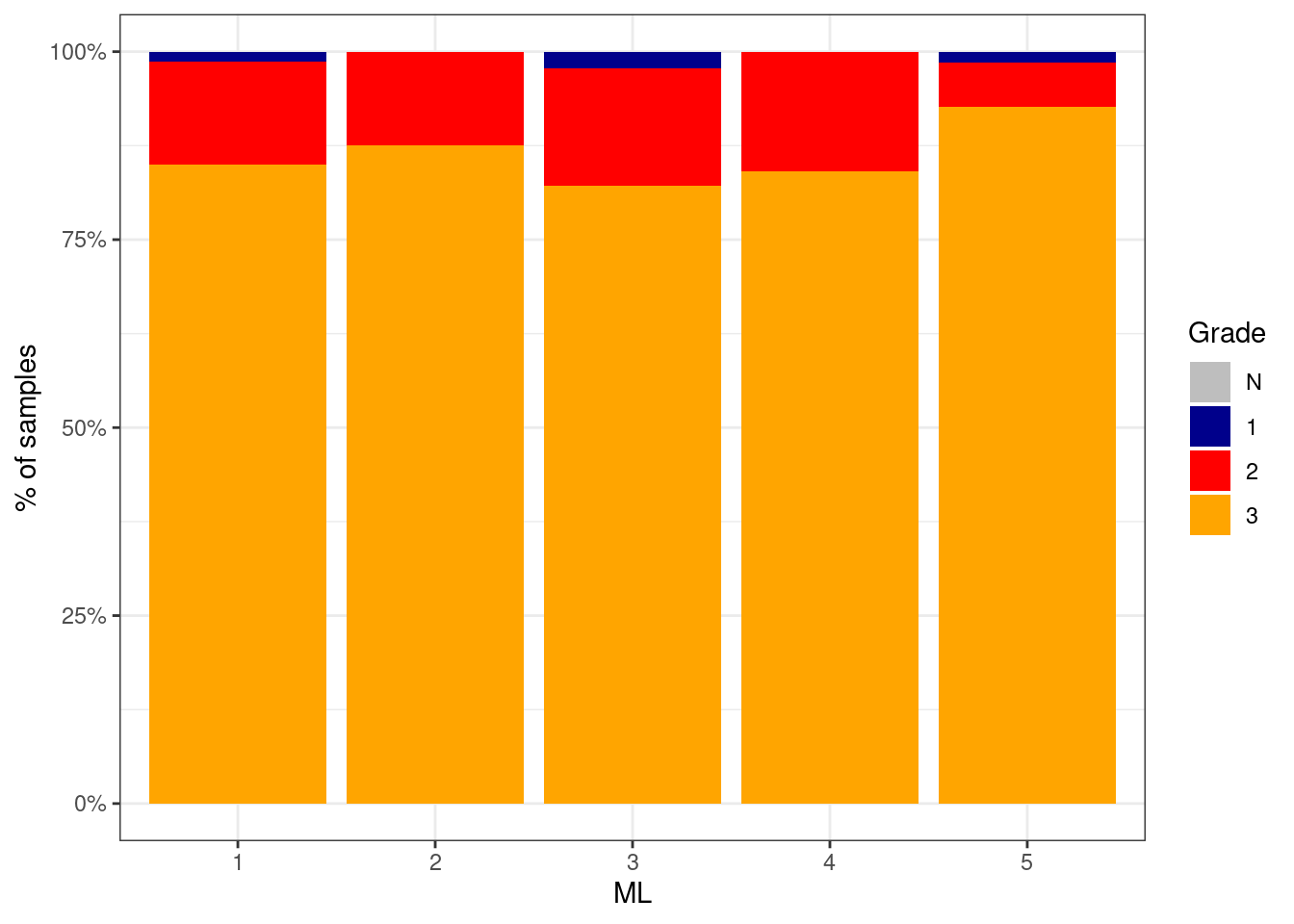
options(repr.plot.width = 6, repr.plot.height = 4)
p_stage_MG_neg <- df %>%
filter(ER == "ER-") %>%
mutate(stage = factor(stage, levels = c("N", "0-1", "2", "3", "4"))) %>%
filter(!is.na(stage)) %>%
count(stage, MG) %>%
group_by(MG) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=MG, y=p, fill=stage)) +
geom_col() +
scale_fill_manual(name = "Stage", values = c("N" = "gray", "0-1" = "black", "2" = "blue", "3" = "red", "4" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_stage_ML_neg <- df %>%
filter(ER == "ER-") %>%
mutate(stage = factor(stage, levels = c("N", "0-1", "2", "3", "4"))) %>%
filter(!is.na(stage)) %>%
count(stage, ML) %>%
group_by(ML) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=ML, y=p, fill=stage)) +
geom_col() +
scale_fill_manual(name = "Stage", values = c("N" = "gray", "0-1" = "black", "2" = "blue", "3" = "red", "4" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_stage_MG_neg + theme_bw()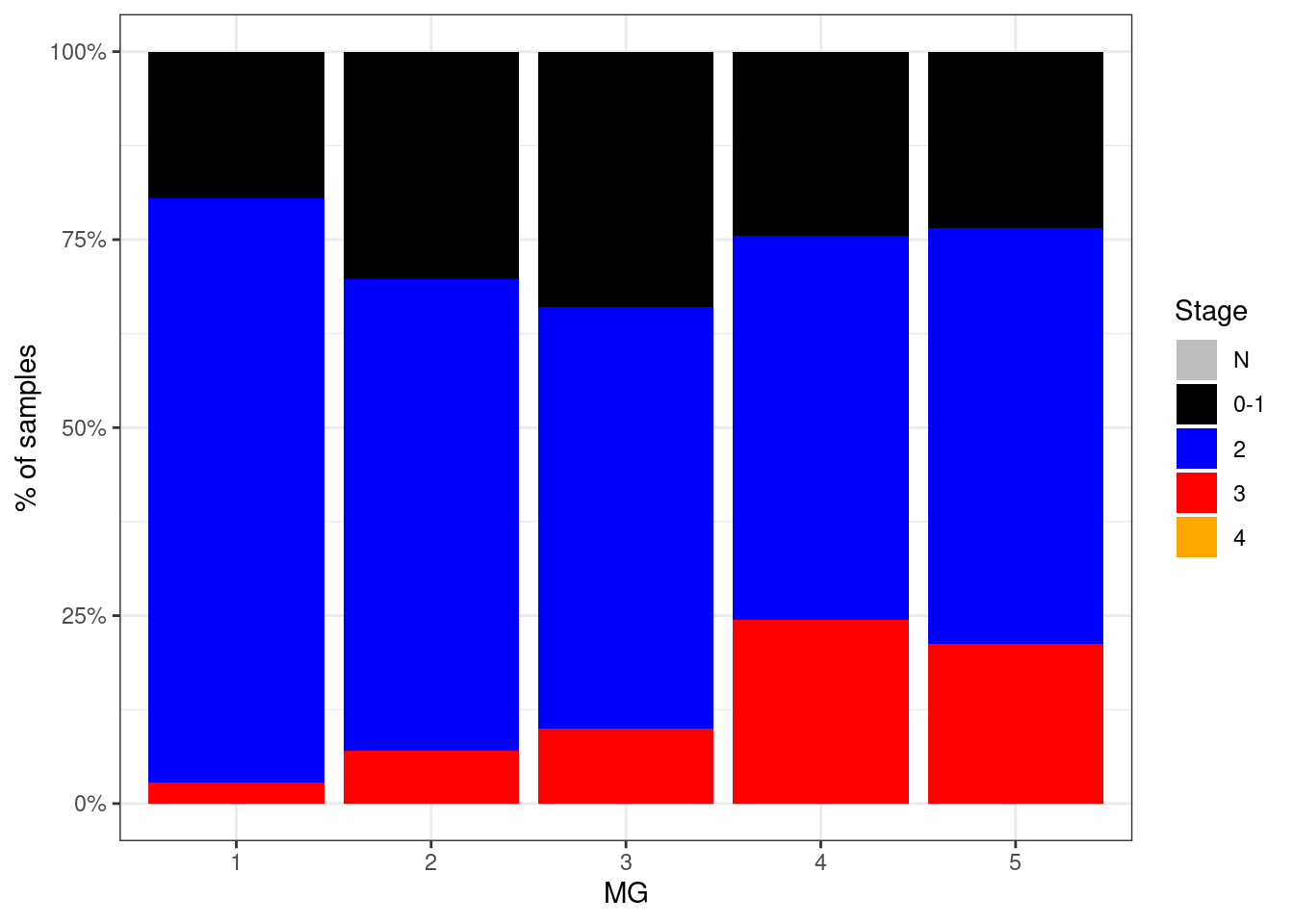
p_stage_ML_neg + theme_bw()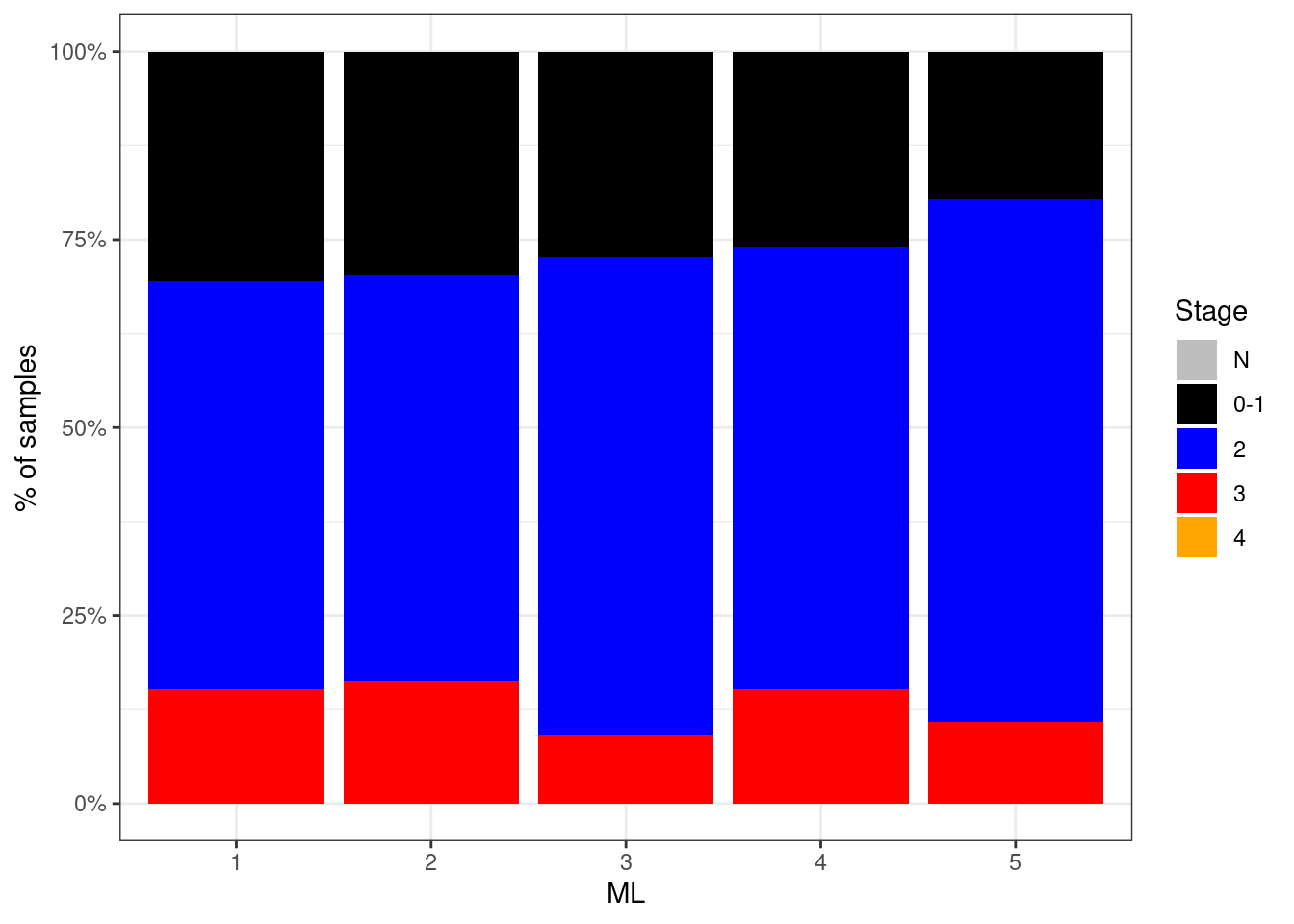
6.2.0.2 Figure 2D
options(repr.plot.width = 6, repr.plot.height = 4)
p_grade_MG <- df %>%
filter(ER == "ER+") %>%
mutate(grade = factor(grade, levels = c("N", "1", "2", "3"))) %>%
filter(!is.na(grade)) %>%
count(grade, MG) %>%
group_by(MG) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=MG, y=p, fill=grade)) +
geom_col() +
scale_fill_manual(name = "Grade", values = c("N" = "gray", "1" = "darkblue", "2" = "red", "3" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_grade_ML <- df %>%
filter(ER == "ER+") %>%
mutate(grade = factor(grade, levels = c("N", "1", "2", "3"))) %>%
filter(!is.na(grade)) %>%
count(grade, ML) %>%
group_by(ML) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=ML, y=p, fill=grade)) +
geom_col() +
scale_fill_manual(name = "Grade", values = c("N" = "gray", "1" = "darkblue", "2" = "red", "3" = "orange")) +
scale_y_continuous(labels=scales::percent) +
ylab("% of samples")
p_grade_MG + theme_bw()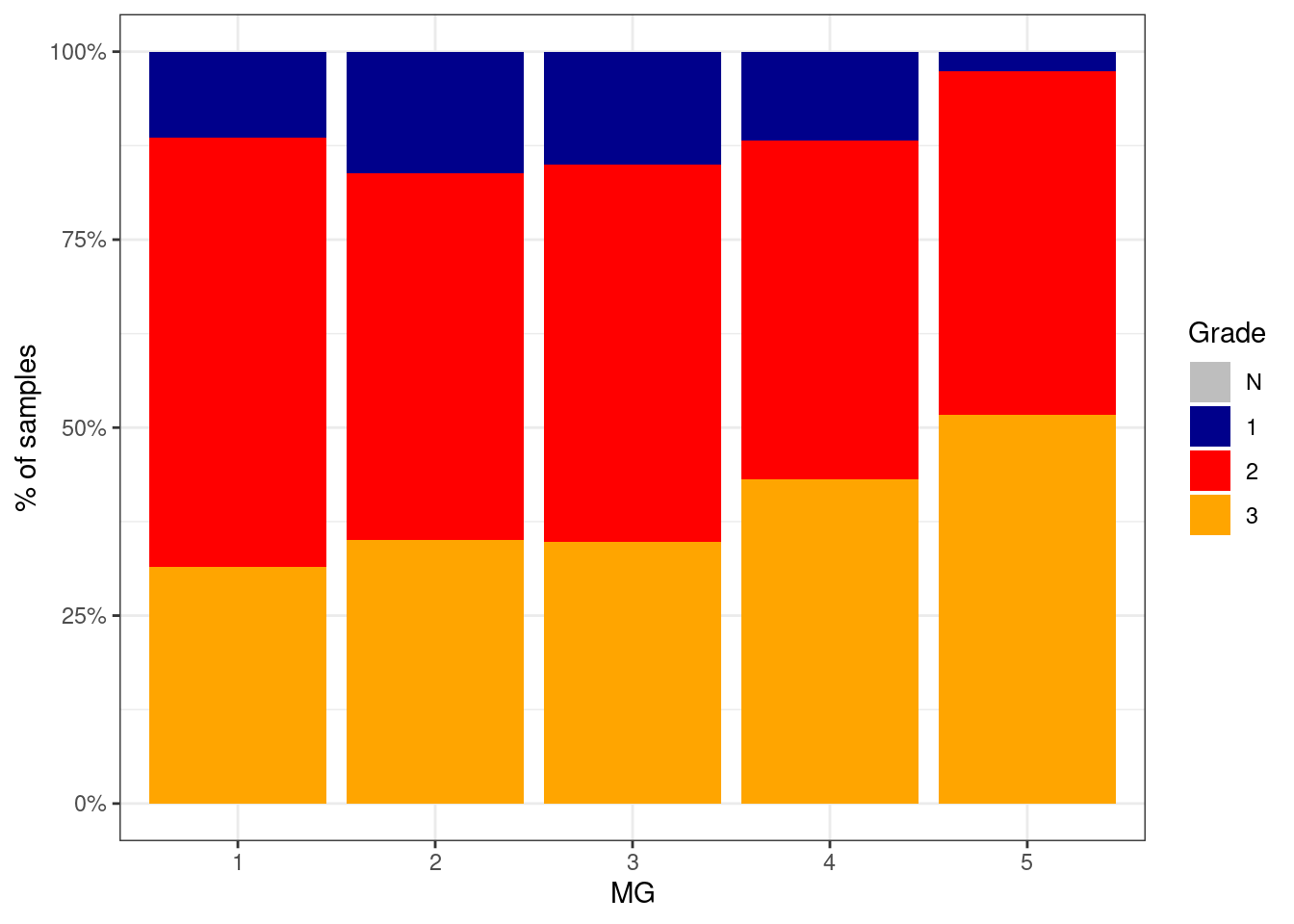
p_grade_ML + theme_bw()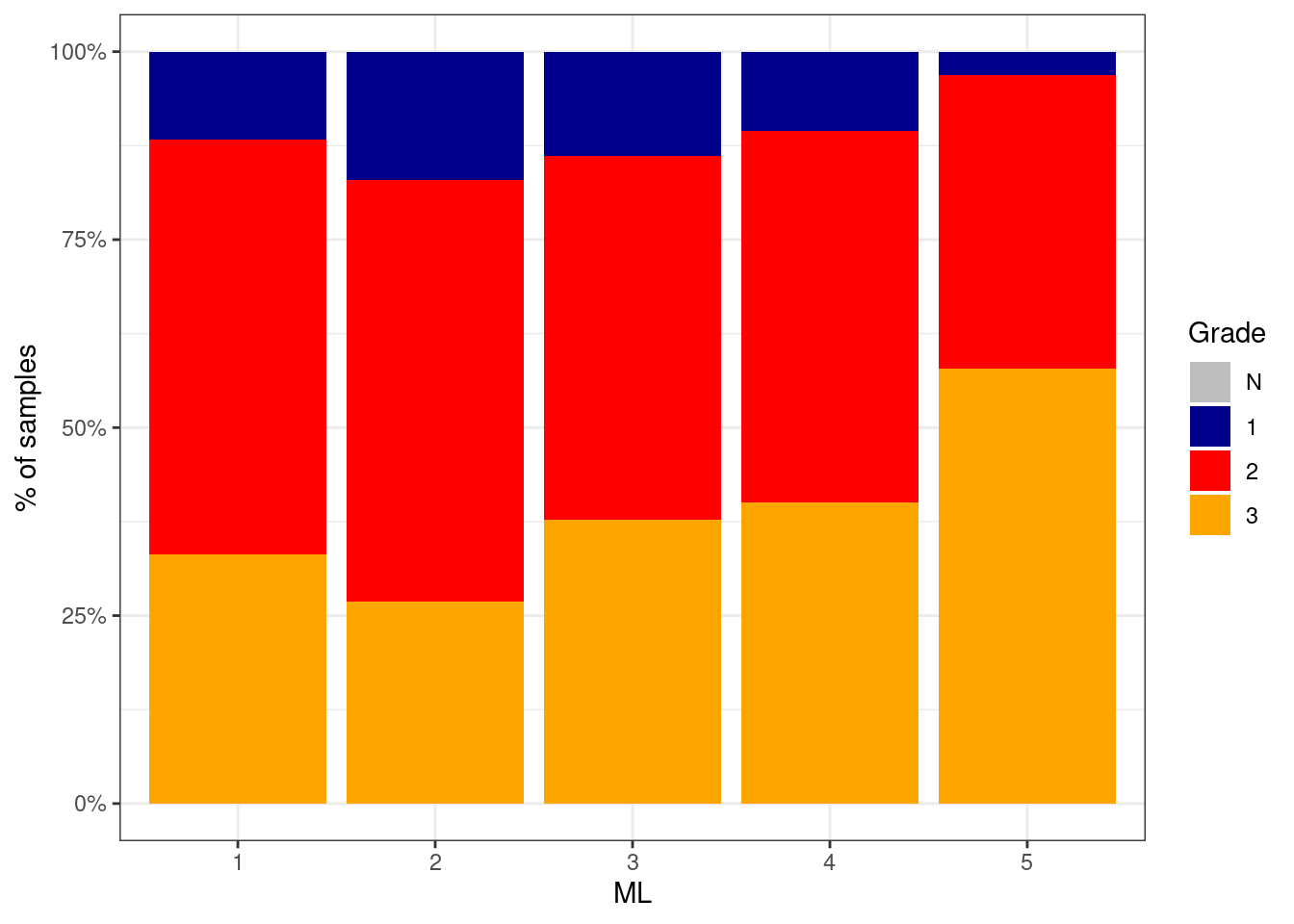
6.2.1 Integrative clusters
6.2.1.1 Extended Data Figure 7C
options(repr.plot.width = 8, repr.plot.height = 6)
df <- feats %>%
left_join(samp_data %>% select(samp, iC10)) %>%
filter(!is.na(iC10)) %>%
mutate(iC10 = factor(iC10, levels = names(annot_colors$iC10))) %>%
select(samp, ER, MG, ML, iC10) %>%
gather("clust", "score", -ER, -iC10, -samp)## Joining, by = "samp"p_iC10 <- df %>%
ggplot(aes(x = iC10, y = score, fill = iC10)) +
geom_boxplot(lwd = 0.2, outlier.size = 0.2) +
facet_grid(clust ~ .) +
guides(fill = FALSE) +
scale_fill_manual(values = annot_colors$iC10) +
ylab("") +
theme(aspect.ratio = 0.5) +
vertical_labs()## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
## "none")` instead.p_iC10 + theme_bw() + theme(aspect.ratio = 0.5) + vertical_labs() + ggpubr::stat_compare_means(method = "kruskal")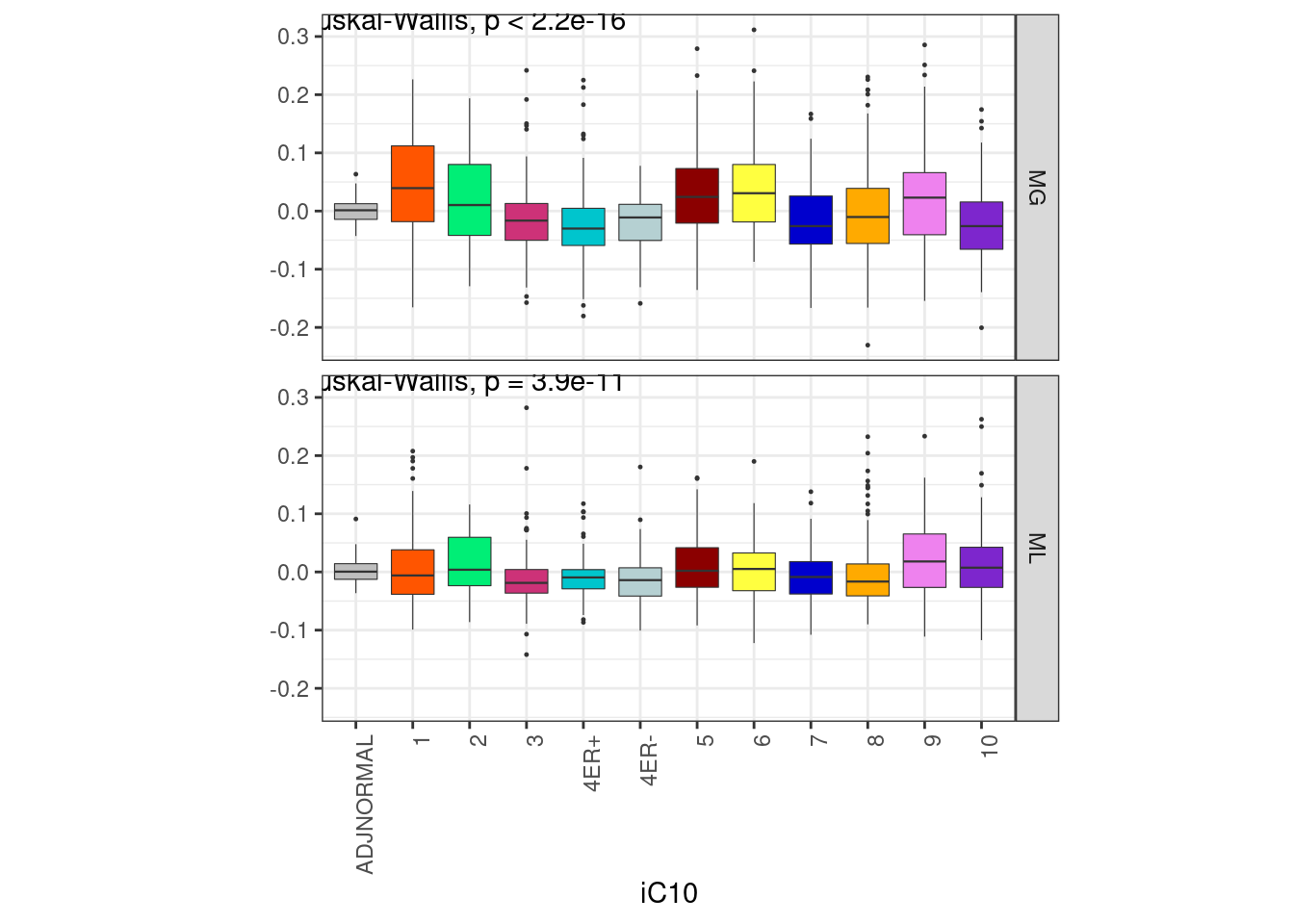
df %>% distinct(samp, iC10) %>% count(iC10)## # A tibble: 12 x 2
## iC10 n
## 1 ADJNORMAL 92
## 2 1 98
## 3 2 54
## 4 3 214
## 5 4ER+ 172
## 6 4ER- 50
## # ... with 6 more rows6.3 Cross correlation (methylation-expression) of epigenomic instability loci
Create a matrix with methylation of loci that are part of MG or ML epigenmic instability and cross correlate it with gene expression in ER+:
all_norm_meth <- fread(here("data/all_norm_meth.tsv")) %>% as_tibble()
ER_positive_mat <- all_norm_meth %>% select(chrom:end, any_of(ER_positive_samples)) %>% intervs_to_mat()
loci_annot <- fread(here("data/loci_annot_epigenomic_features.tsv")) %>% as_tibble()
cor_thresh <- 0.3coords <- loci_annot %>%
filter(abs(MG) >= cor_thresh | abs(ML) >= cor_thresh & abs(clock) < cor_thresh) %>%
select(chrom:end) %>% intervs_to_mat() %>% rownames()
meth_mat <- ER_positive_mat[coords, ]
expr_m <- fread(here("data/expression_matrix.csv")) %>% select(-any_of(c("chrom", "start", "end", "name3.chr")))
expr_mat <- expr_m %>%
as.data.frame() %>%
column_to_rownames("name")
f <- rowSums(!is.na(expr_mat)) > 0
expr_mat <- expr_mat[f, ]em_cross <- em_cross_cor(meth_mat, expr_mat, meth_cor_thresh = 0.25, expr_cor_thresh = 0.25) %cache_rds% here("data/MG_ML_em_cross_cor.rds")em_cross_clust <- cluster_em_cross_cor(em_cross, k_meth = 32, k_expr = 32) %cache_rds% here("data/MG_ML_em_cross_cor_clust.rds")6.3.0.1 Figure 2F
options(repr.plot.width = 8, repr.plot.height = 13)
plot_em_cross_cor(em_cross_clust)## plotting em cross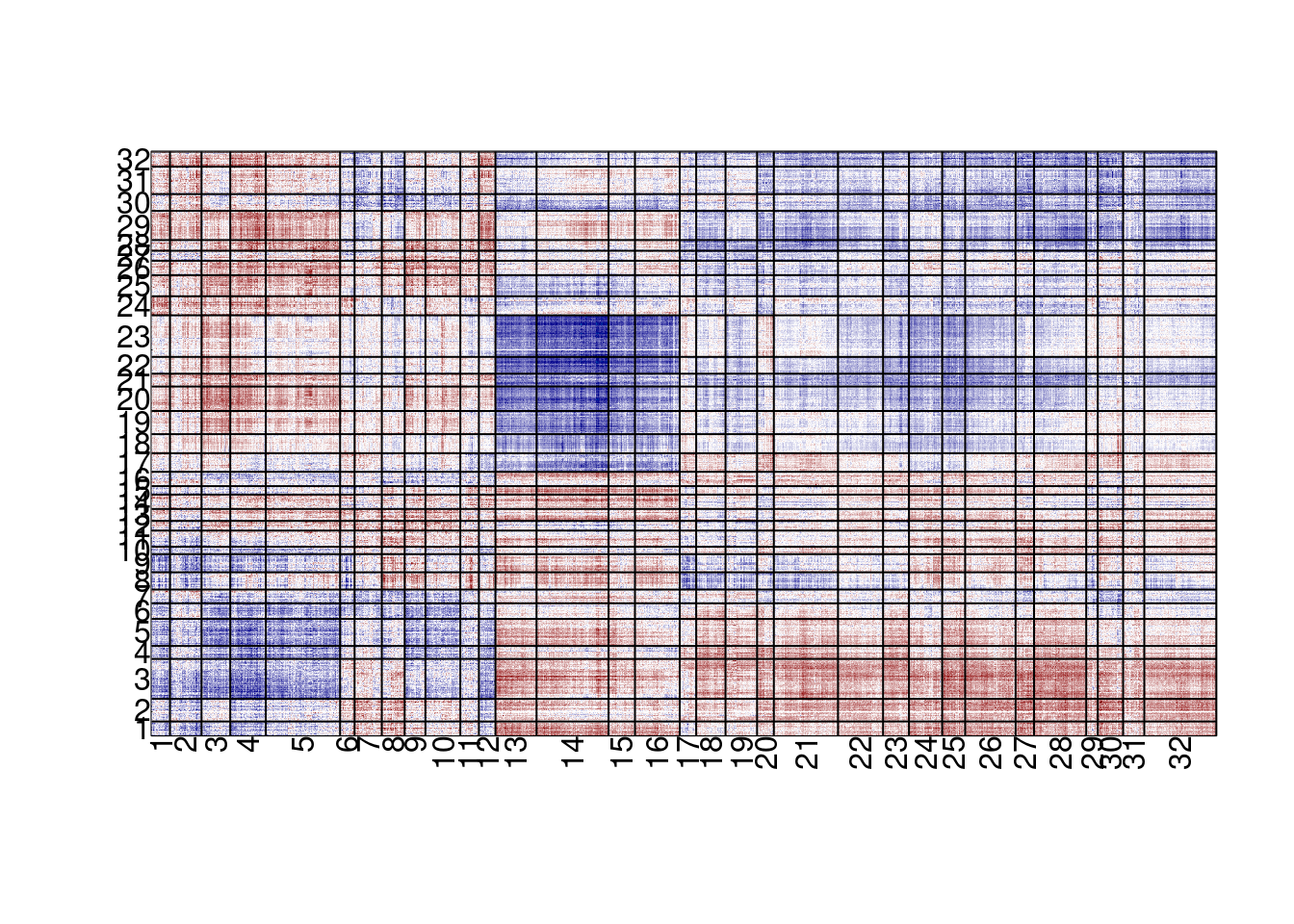
Annotating the methylation clusters we can see which are MG and which are ML
options(repr.plot.width = 10, repr.plot.height = 7)
cutree_order(em_cross_clust$hc_meth, k=32) %>%
mat_to_intervs() %>%
set_names(c("chrom", "start", "end", "clust")) %>%
left_join(loci_annot %>% select(chrom, start, end,clock, MG, ML)) %>%
mutate(feat = case_when(
clock >= cor_thresh ~ "clock",
MG >= cor_thresh ~ "MG",
ML >= cor_thresh ~ "ML",
TRUE ~ "other")) %>%
count(clust, feat) %>%
as_tibble() %>%
group_by(clust) %>%
mutate(p = n / sum(n)) %>%
ggplot(aes(x=factor(clust), y=n, fill=feat)) + geom_col() + theme_bw()## Joining, by = c("chrom", "start", "end")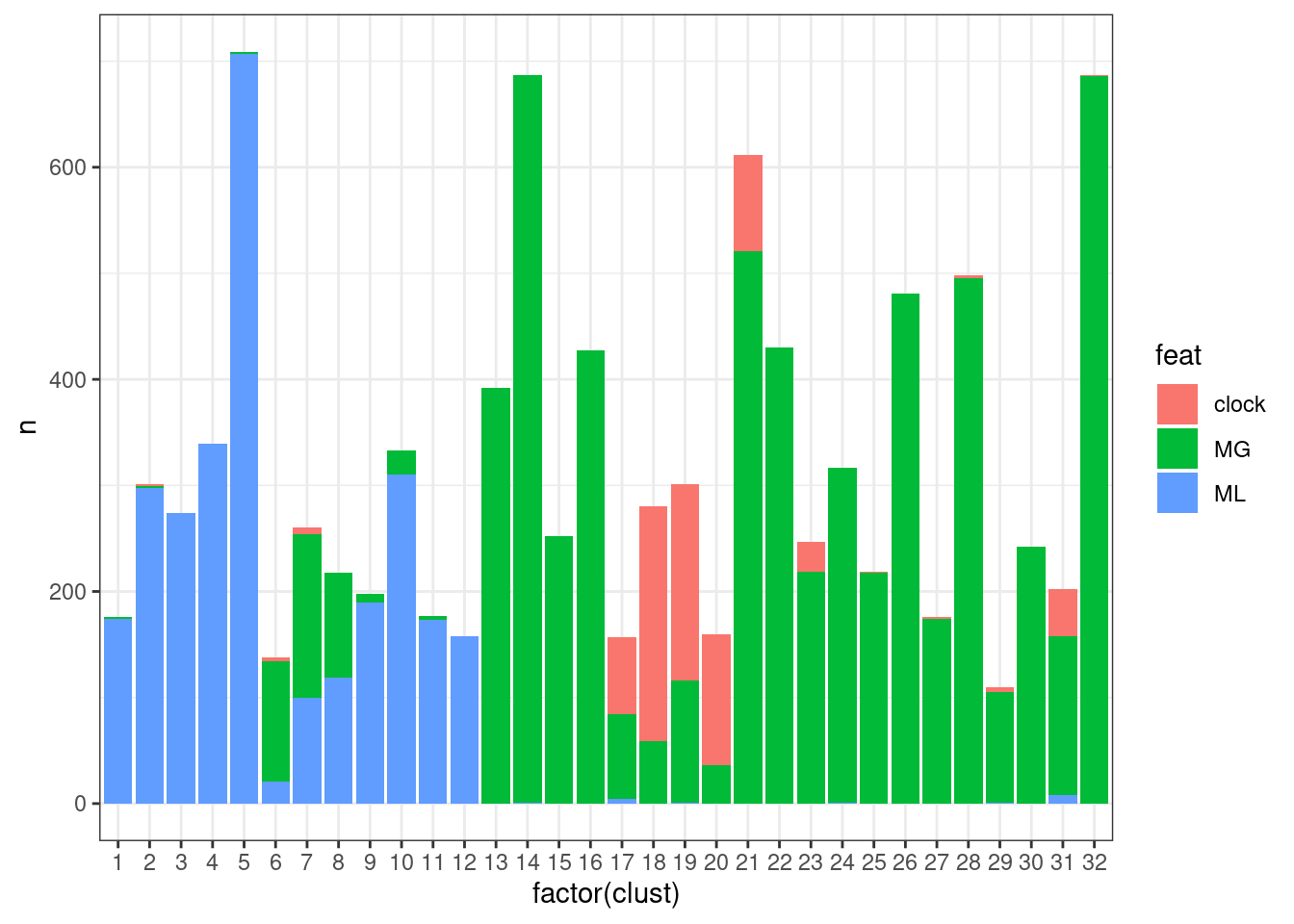
options(repr.plot.width = 8, repr.plot.height = 13)
plot_em_cross_cor(em_cross_clust, k_meth = 3, k_exp = 32)## plotting em cross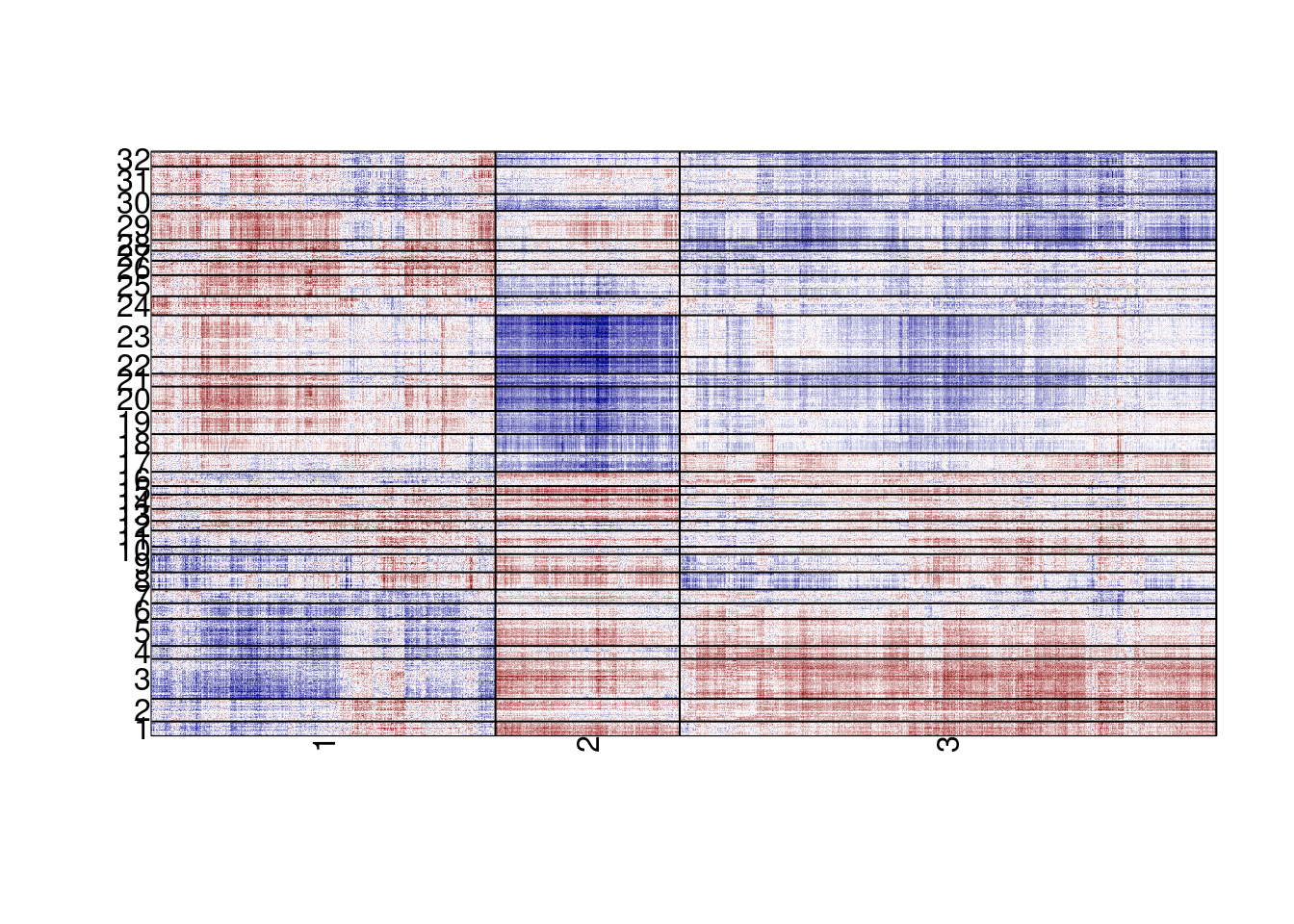
em_cross_clust$expr_clust %>% arrange(clust) %>% fwrite(here("data/MG_ML_em_cross_gene_clust_id.tsv"), sep="\t")Plot for each expression cluster the name of the gene with highest correlation to any locus.
top_genes <- em_cross_clust$em_cross %>%
gather_matrix("name", "locus", "cor") %>%
left_join(em_cross_clust$expr_clust) %>%
arrange(clust, cor) %>%
group_by(clust) %>%
slice(1) %>%
ungroup() %>%
left_join(em_cross_clust$expr_clust %>%
count(clust) %>%
mutate(p = cumsum(n / sum(n)), pos = p - (p - lag(p, default = 0)) / 2) %>%
select(clust, pos)) %cache_df% here("data/MG_ML_em_cross_top_genes.tsv")p_gene_names <- ggplot(top_genes, aes(x = 1, y = pos, label = name)) + geom_text() + theme_void(base_size = 3, base_family = "Arial")Plot barplots of MG gene expression correlations
gene_annots <- get_gene_annots()## Joining, by = "type"feat_gene_cors <- get_expression_features_cors()gene_annots_cors <- feat_gene_cors %>% left_join(gene_annots, by = "name") %>% filter(ER == "ER+", MG >= 0.15) %>% distinct() 6.3.0.2 Figure 2G
options(repr.plot.width = 8, repr.plot.height = 8)
p_cc <- gene_annots_cors %>%
filter(type == "Cell Cycle") %>%
ggplot(aes(x = reorder(name, MG), y = MG)) + geom_col(fill = "#008B45FF") + xlab("") + ylab("Correlation to MG") + vertical_labs() + ggtitle("Cell Cycle") + ylim(0, 0.5) + theme(axis.text.x = element_text(size = 4, family = "Arial")) + theme(plot.title = element_text(hjust = 0.5), panel.grid.major.x = element_blank())
p_emb <- gene_annots_cors %>%
filter(type == "Embryonic TF") %>%
ggplot(aes(x = reorder(name, MG), y = MG)) + geom_col(fill = "#3B4992FF") + xlab("") + ylab("Correlation to MG") + vertical_labs() + ggtitle("Embryonic TF") + ylim(0, 0.5) + theme(axis.text.x = element_text(size = 4, family = "Arial"), plot.title = element_text(hjust = 0.5), panel.grid.major.x = element_blank())
p_other <- gene_annots_cors %>%
filter(type == "Other") %>%
ggplot(aes(x = reorder(name, MG), y = MG)) + geom_col(fill = "black") + xlab("") + ylab("Correlation to MG") + vertical_labs() + ggtitle("Other") + ylim(0, 0.5) + theme(axis.text.x = element_text(size = 4, family = "Arial"), plot.title = element_text(hjust = 0.5), panel.grid.major.x = element_blank())
p_cc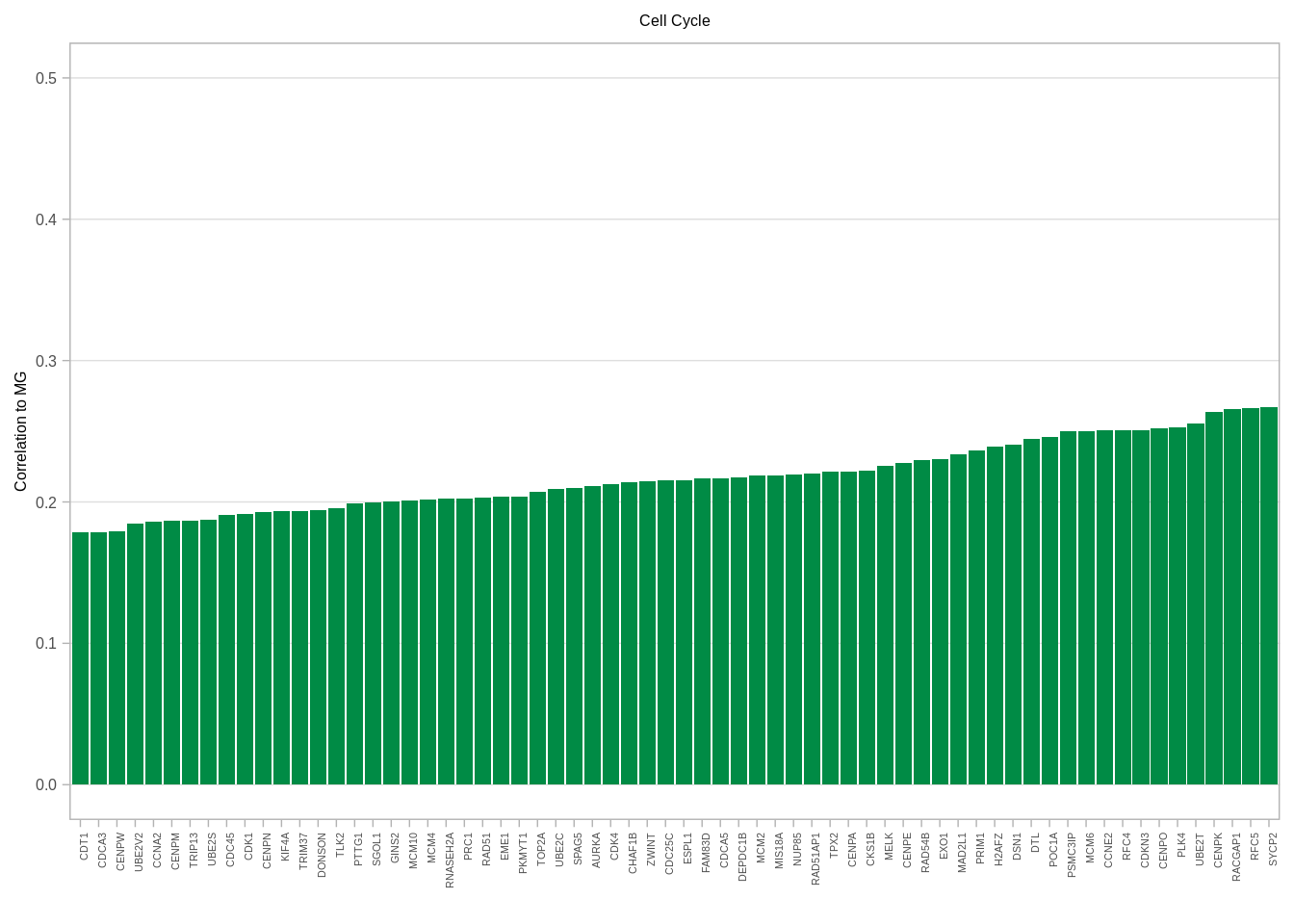
p_emb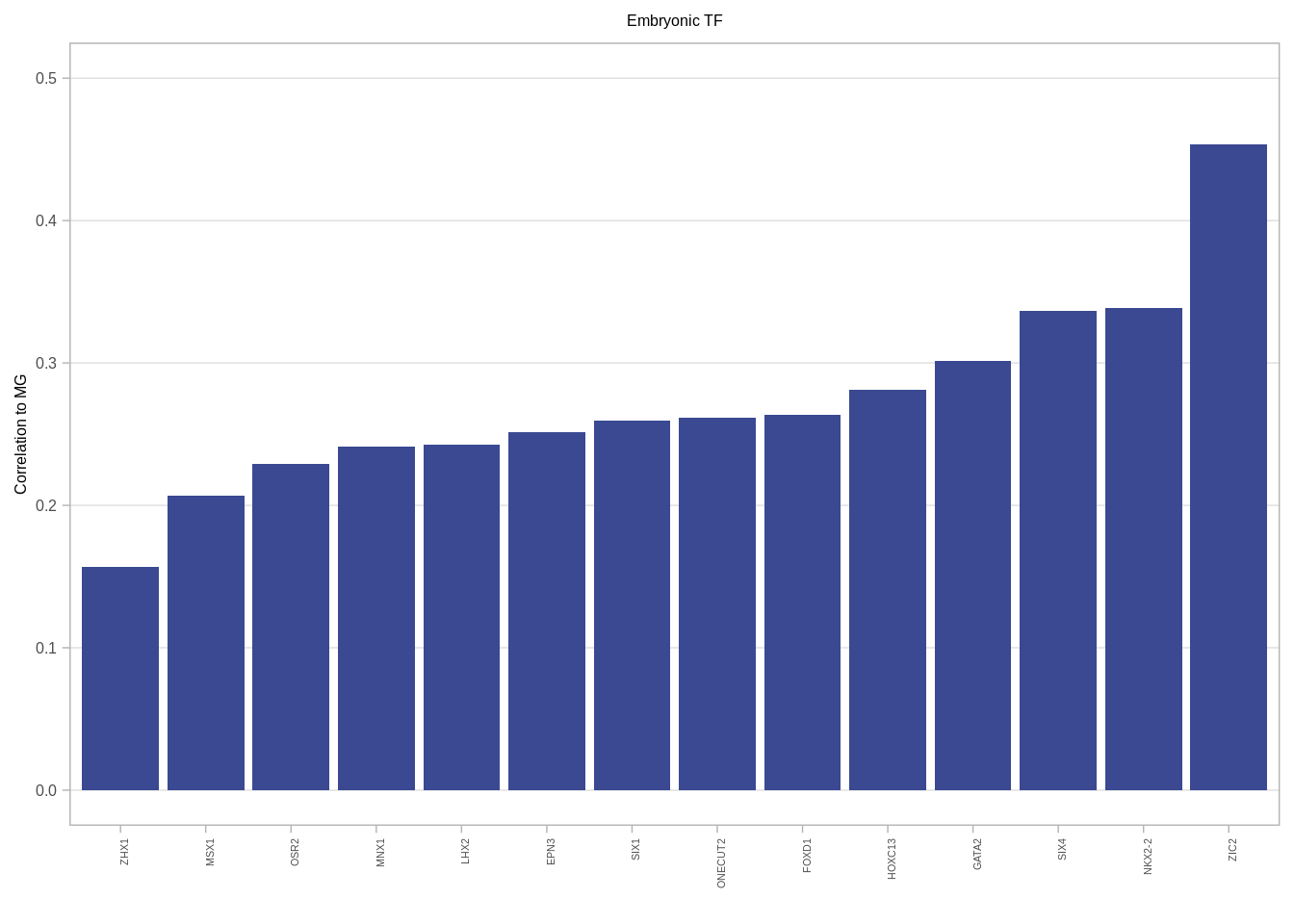
p_other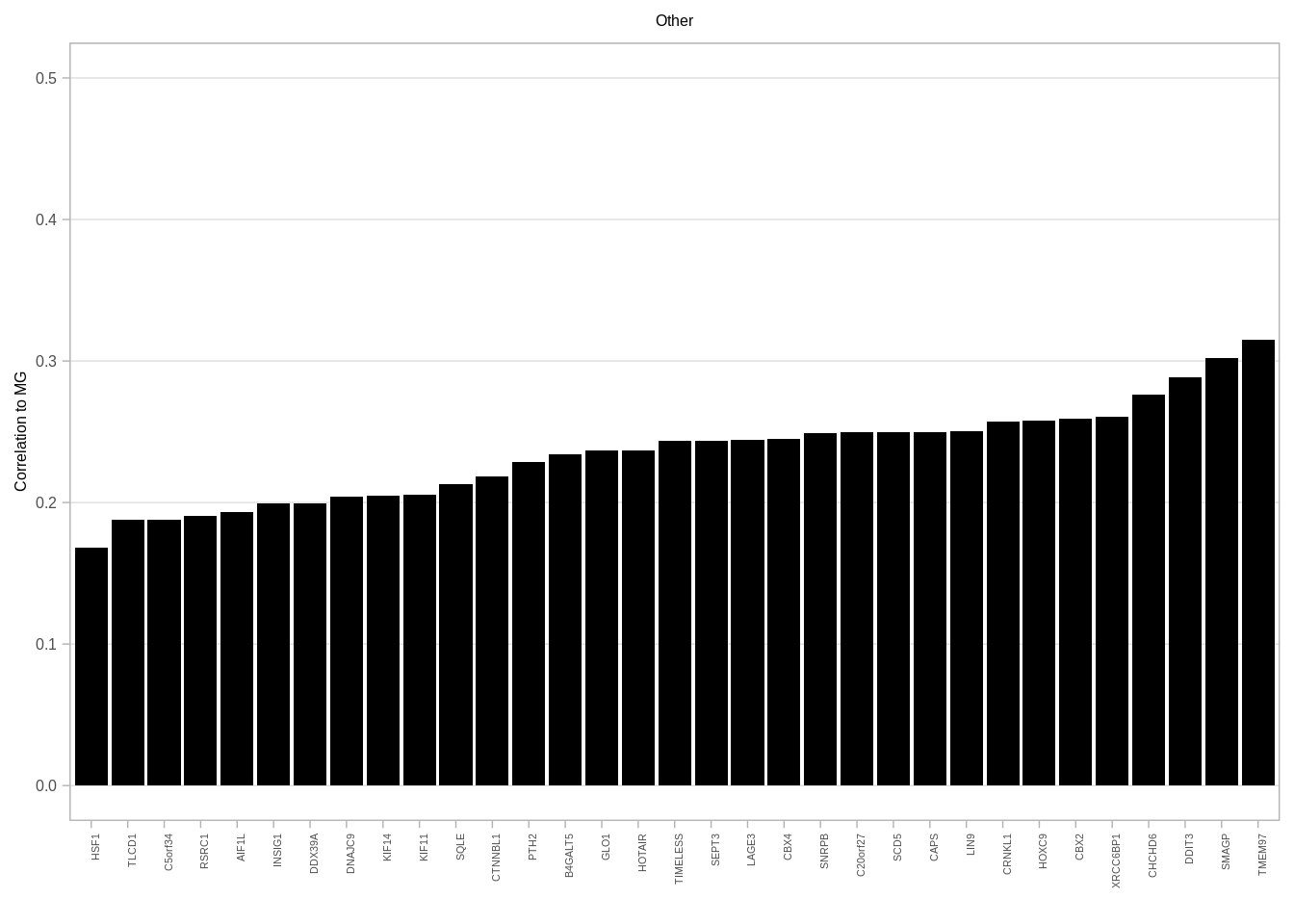
6.4 Epigenomic instability in different Time of replication (TOR) regimes
loci_annot <- fread(here("data/loci_annot_epigenomic_features.tsv")) %>% as_tibble()
loci_clust <- fread(here("data/ER_positive_loci_clust_df.tsv")) %>% as_tibble()
loci_clust <- loci_clust %>% left_join(loci_annot)## Joining, by = c("chrom", "start", "end")loci_clust_MG <- loci_clust %>% filter(clust == "MG")
loci_clust_ML <- loci_clust %>% filter(clust == "ML")all_mat_raw <- get_all_meth() %>% intervs_to_mat()tor_strata <- loci_clust_MG %>% mutate(tor_strata = cut(tor, breaks = main_config$genomic_regions$tor_low_mid_high, labels=c("late", "intermediate", "early"))) %>% pull(tor_strata) %>% forcats::fct_explicit_na()
intervs <- loci_clust_MG %>% intervs_to_mat() %>% rownames()samp_meth_tor_MG <- tgs_matrix_tapply(t(all_mat_raw[intervs, ]), tor_strata, mean, na.rm=TRUE) %>% t() %>% as.data.frame() %>% rownames_to_column("samp") %>% add_ER() %>% as_tibble()6.4.0.1 Extended Data Figure 7B
options(repr.plot.width = 4, repr.plot.height = 4)
p_early_late <- samp_meth_tor_MG %>%
filter(ER != "normal") %>%
ggplot(aes(x=early, y=late, color=ER)) +
geom_abline(linetype = "dashed") +
geom_point(size=0.5) +
xlim(0, 0.8) +
ylim(0, 0.8) +
xlab("MG loci: early replicating") +
ylab("MG loci: late replicating") +
scale_color_manual(values=annot_colors$ER1) +
theme(aspect.ratio = 1)
samp_meth_tor_MG %>% filter(ER != "normal") %>% summarise(cor_early_late = cor(early, late, method = "spearman", use = "pairwise.complete.obs"), cor_early_mid = cor(early, intermediate, method = "spearman", use = "pairwise.complete.obs"))## # A tibble: 1 x 2
## cor_early_late cor_early_mid
## 1 0.8602804 0.9382058p_early_late + theme_bw() + theme(aspect.ratio = 1) 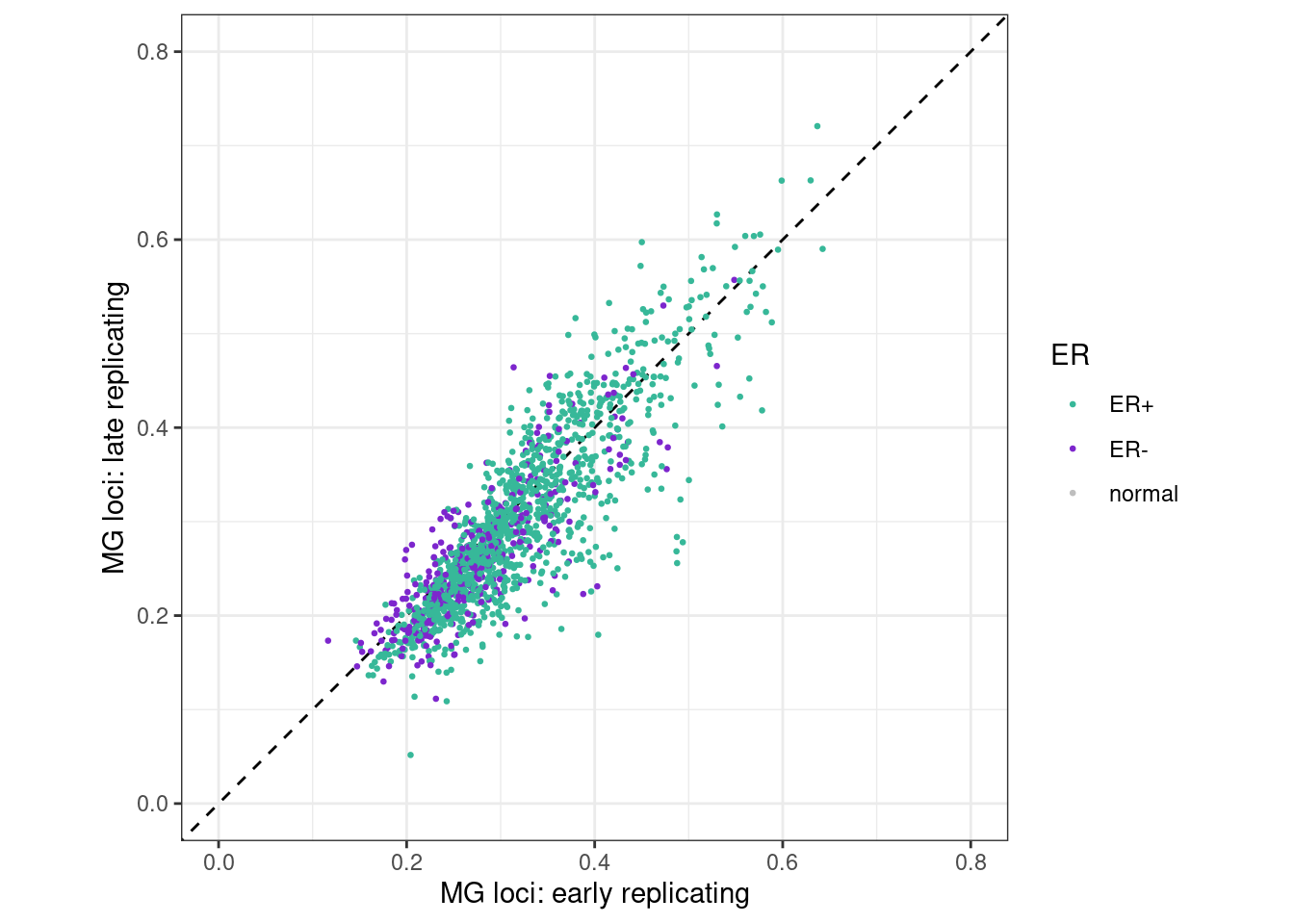
tor_strata <- loci_clust_ML %>% mutate(tor_strata = cut(tor, breaks = main_config$genomic_regions$tor_low_mid_high, labels=c("late", "intermediate", "early"))) %>% pull(tor_strata) %>% forcats::fct_explicit_na()
intervs <- loci_clust_ML %>% intervs_to_mat() %>% rownames()samp_meth_tor_ML <- tgs_matrix_tapply(t(all_mat_raw[intervs, ]), tor_strata, mean, na.rm=TRUE) %>% t() %>% as.data.frame() %>% rownames_to_column("samp") %>% add_ER() %>% as_tibble()options(repr.plot.width = 4, repr.plot.height = 4)
p_early_late_ML <- samp_meth_tor_ML %>%
filter(ER != "normal") %>%
ggplot(aes(x=early, y=late, color=ER)) +
geom_abline(linetype = "dashed") +
geom_point(size=0.5) +
xlim(0, 0.8) +
ylim(0, 0.8) +
xlab("ML loci: early replicating") +
ylab("ML loci: late replicating") +
scale_color_manual(values=annot_colors$ER1) +
theme(aspect.ratio = 1)
samp_meth_tor_ML %>% filter(ER != "normal") %>% summarise(cor_early_late = cor(early, late, method = "spearman", use = "pairwise.complete.obs"), cor_early_mid = cor(early, intermediate, method = "spearman", use = "pairwise.complete.obs"))## # A tibble: 1 x 2
## cor_early_late cor_early_mid
## 1 0.8201663 0.9276362p_early_late_ML + theme_bw() + theme(aspect.ratio = 1) ## Warning: Removed 32 rows containing missing values (geom_point).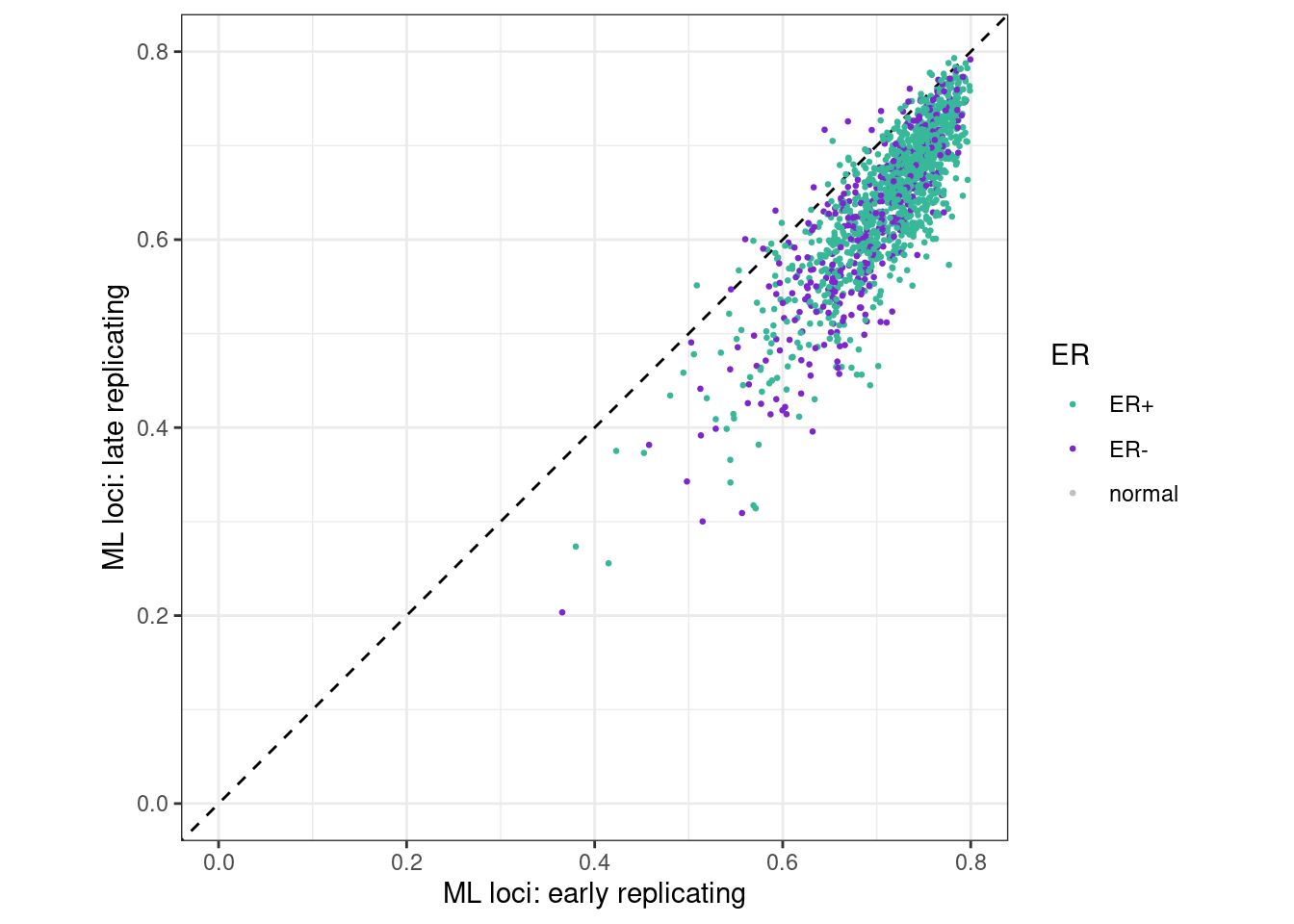
gc()## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 4690952 250.6 8042191 429.5 8042191 429.5
## Vcells 1417529451 10814.9 2192953529 16731.0 1803628477 13760.6