8 Mutations
source(here::here("scripts/init.R"))mut_df <- get_mut_df()
dim(mut_df)## [1] 283689 4mut_df%>% distinct(samp) %>% nrow()## [1] 1659We compare the distribution of the epigenomic scores with and without mutation for a panel of 171 genes. We do it separatly for activating (+) and inactivating (-) mutations.
calc_features_mutation_pvals <- function(min_n = 10) {
feats_tidy <- get_all_features() %>%
select(-caf, -immune) %>%
gather("feat", "score", -ER, -samp)
mut_df <- get_mut_df()
df <- mut_df %>% left_join(feats_tidy, by = "samp")
pvals_minus <- df %>%
group_by(gene, ER, feat) %>%
filter(sum(!is.na(score[mutation == "MUT-"])) >= min_n) %>%
summarise(pval = wilcox.test(score[mutation == "MUT-"], score[mutation %in% c("NO MUT", "MUT+")])$p.value) %>%
ungroup() %>%
mutate(type = "MUT-")
pvals_plus <- df %>%
group_by(gene, ER, feat) %>%
filter(sum(!is.na(score[mutation == "MUT+"])) >= min_n) %>%
summarise(pval = wilcox.test(score[mutation == "MUT+"], score[mutation %in% c("NO MUT", "MUT-")])$p.value) %>%
ungroup() %>%
mutate(type = "MUT+")
pvals <- bind_rows(pvals_minus, pvals_plus) %>%
group_by(ER) %>%
mutate(qval = p.adjust(pval))
return(pvals)
} %cache_df% "data/features_mutations_pvals.tsv"
pvals <- calc_features_mutation_pvals()signif_genes <- pvals %>%
filter(qval <= 0.01) %>%
pull(gene) %>%
unique()
pvals %>% filter(qval <= 0.01) %>% arrange(gene) %>% mutate(signif = case_when(qval < 0.0001 ~ "****", qval < 0.001 ~ "***", qval < 0.01 ~ "**", qval < 0.05 ~ "*"))## # A tibble: 6 x 7
## # groups: ER
## gene ER feat pval type qval signif
## 1 CBFB ER+ ML 1.859177e-06 MUT- 2.658623e-04 ***
## 2 CDH1 ER+ MG 1.250775e-07 MUT- 1.801116e-05 ****
## 3 GATA3 ER+ ML 3.488426e-05 MUT- 4.918680e-03 **
## 4 PIK3CA ER- MG 6.510399e-07 MUT+ 1.302080e-05 ****
## 5 TP53 ER+ immune.meth 4.098744e-06 MUT- 5.820217e-04 ***
## 6 TP53 ER+ ML 4.633858e-20 MUT- 6.719094e-18 ****We plot the distribution of scores and mutations for the genes that were significant.
8.0.0.1 Figure 4B
options(repr.plot.width = 10, repr.plot.height = 10)
feats_tidy <- get_all_features() %>%
select(-caf, -immune) %>%
gather("feat", "score", -ER, -samp)
mut_df_count <- get_mut_df() %>%
left_join(feats_tidy) %>%
mutate(mutation = factor(mutation, levels = c("NO MUT", "MUT-", "MUT+", "NOR"))) %>%
filter(ER != "normal", mutation != "") %>%
group_by(feat, ER) %>%
mutate(score = cut(score, breaks = quantile(score, 0:5 / 5, na.rm = TRUE), include.lowest = TRUE, labels = as.character(1:5))) %>%
count(gene, ER, feat, mutation, score) %>%
group_by(gene, ER, feat, score) %>%
mutate(p = n / sum(n)) %>%
ungroup()## Joining, by = "samp"p <- mut_df_count %>%
filter((gene == "PIK3CA" & ER == "ER-") | (gene != "PIK3CA" & ER == "ER+"), gene %in% signif_genes) %>%
mutate(feat = factor(feat, c("caf.meth", "immune.meth", "clock", "MG", "ML"))) %>%
mutate(gene = paste0(gene, " (", ER, ")")) %>%
ggplot(aes(fill = mutation, y = p, x = score, color = mutation, label = n)) +
geom_col(width = 1, color = "black") +
scale_color_manual(values = c("MUT+" = "white", "MUT-" = "white", "NO MUT" = "black")) +
scale_fill_manual(values = annot_colors$mutation) +
geom_text(family = "Arial", size = 1.5, position = position_stack(vjust = 0.5)) +
guides(color = "none") +
facet_grid(feat ~ gene) +
ylab("% of samples") +
xlab("Score") +
scale_y_continuous(labels = scales::percent) +
theme(
aspect.ratio = 0.7,
axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
panel.border = element_blank()
)
p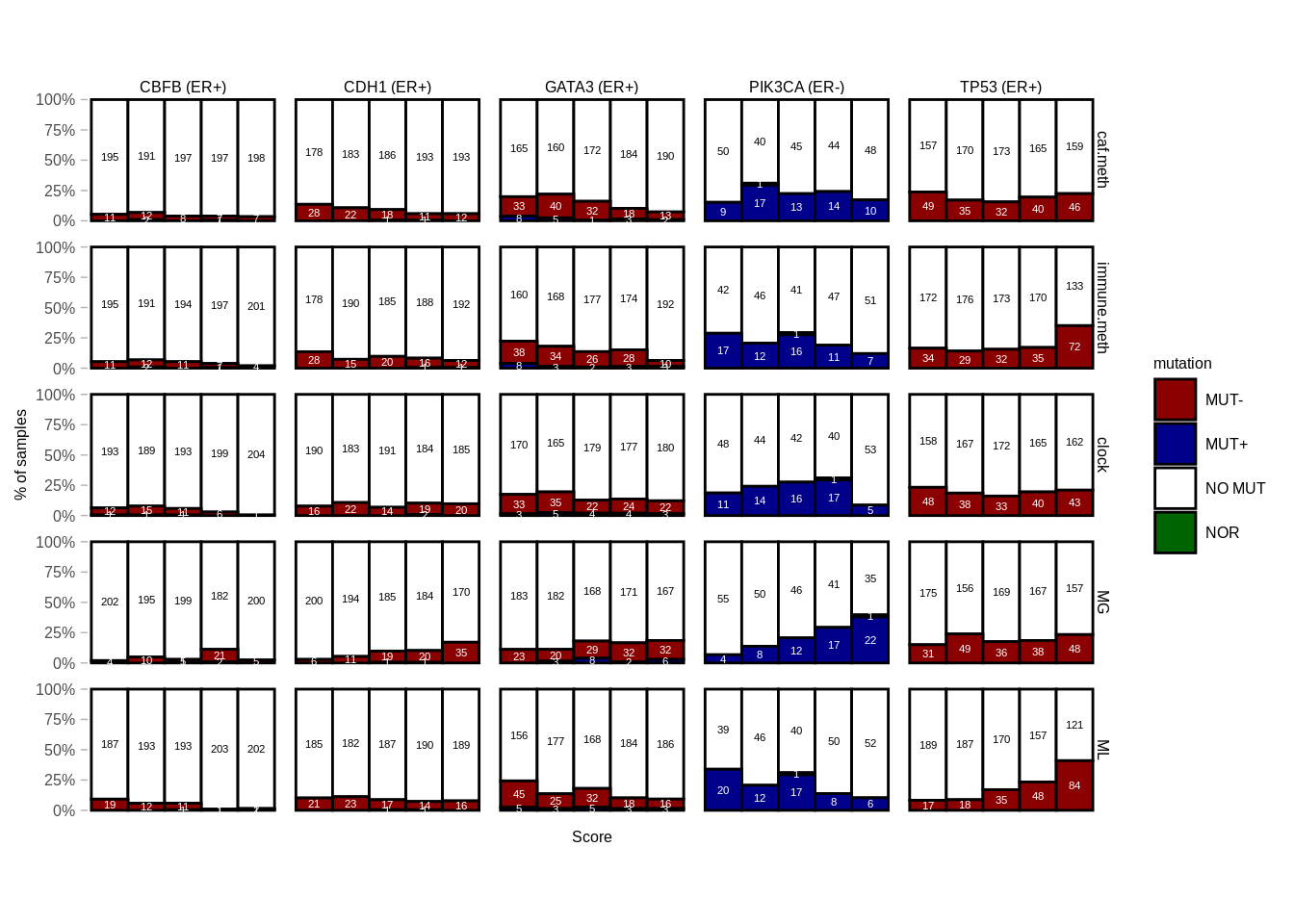
We can test which associations hold even after adding PAM50 as a confounder:
calc_features_mutation_pvals_PAM50 <- function(min_n = 10) {
feats_tidy <- get_all_features() %>%
select(-caf, -immune) %>%
gather("feat", "score", -ER, -samp)
mut_df <- get_mut_df()
df <- mut_df %>% left_join(feats_tidy, by = "samp")
df <- df %>% left_join(samp_data %>% select(samp, PAM50))
pvals_minus <- df %>%
group_by(gene, ER, PAM50, feat) %>%
filter(sum(!is.na(score[mutation == "MUT-"])) >= min_n) %>%
summarise(pval = wilcox.test(score[mutation == "MUT-"], score[mutation %in% c("NO MUT", "MUT+")])$p.value) %>%
ungroup() %>%
mutate(type = "MUT-")
pvals_plus <- df %>%
group_by(gene, ER, PAM50, feat) %>%
filter(sum(!is.na(score[mutation == "MUT+"])) >= min_n) %>%
summarise(pval = wilcox.test(score[mutation == "MUT+"], score[mutation %in% c("NO MUT", "MUT-")])$p.value) %>%
ungroup() %>%
mutate(type = "MUT+")
pvals <- bind_rows(pvals_minus, pvals_plus) %>%
group_by(ER, PAM50) %>%
mutate(qval = p.adjust(pval))
return(pvals)
} %cache_df% "data/features_mutations_pvals_PAM50.tsv"
pvals_pam50 <- calc_features_mutation_pvals_PAM50()## Joining, by = "samp"signif_genes_pam50 <- pvals_pam50 %>%
filter(qval <= 0.01) %>%
pull(gene) %>%
unique()
pvals_pam50 %>% filter(qval <= 0.01) %>% arrange(gene)## # A tibble: 5 x 7
## # groups: ER, PAM50
## gene ER PAM50 feat pval type qval
## 1 CDH1 ER+ LUMA MG 0.0000063740045 MUT- 0.00050354636
## 2 GATA3 ER+ LUMA ML 0.0000465714399 MUT- 0.00363257231
## 3 PIK3CA ER+ LUMA ML 0.0000004288503 MUT+ 0.00003430802
## 4 TP53 ER+ LUMA ML 0.0000823223268 MUT- 0.00633881916
## 5 TP53 ER+ LUMB ML 0.0000016139331 MUT- 0.00008069666gc()## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 4262458 227.7 7549761 403.3 7549761 403.3
## Vcells 13663688 104.3 45292323 345.6 43948331 335.3